
本文作者:杉杉
导读
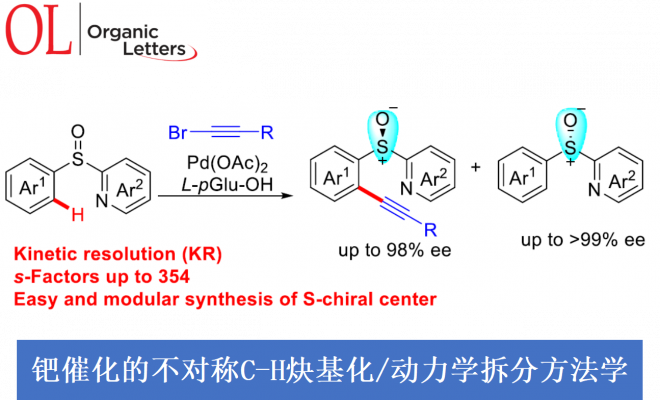
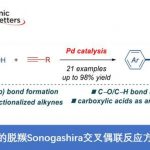
近日,浙江大学的史炳锋与五邑大学的徐学涛等课题组在Org. Lett.中发表论文,报道一种通过钯(II)催化剂参与的2-(芳基亚磺酰基)吡啶 (2‑(Arylsulfinyl)pyridines)的对映选择性C-H炔基化反应方法学。其中,关键步骤涉及采用廉价易得的L-pGlu-OH作为手性配体进行的动力学拆分过程。同时,这一全新的对映选择性C-H炔基化策略具有广泛的底物应用范围以及良好的官能团兼容性等优势,并能够以优良的反应收率与优良的对映选择性 (高达99% ee)分别获得相应的手性炔基化产物以及非活性的手性亚砜原料。此外,研究表明,对映富集的C-H炔基化产物能够较为容易地转化为其它不同类型的手性亚砜砌块,并且,反应过程中,对映纯度未受影响。
Synthesis of Chiral Sulfoxides via Pd(II)-Catalyzed Enantioselective C-H Alkynylation/Kinetic Resolution of 2‑(Arylsulfinyl)pyridines
T. Zhou, Meng. Jiang, P. Qian, Q. Yao, X. Xu, K. Zhang, B. Shi, Org. Lett. ASAP doi: 10.1021/acs.orglett.1c02918.
正文
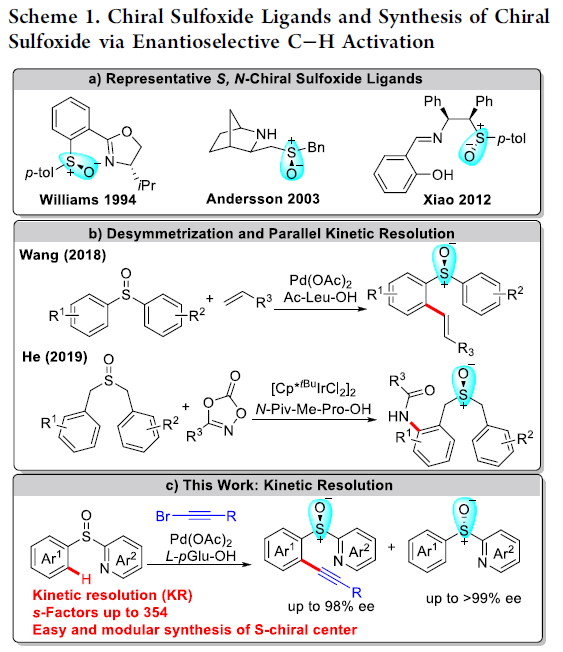
具有手性硫原子的亚砜是诸多药物分子以及生物活性化合物中,普遍存在的结构单元。并且,手性亚砜同样能够作为不对称催化研究中较为常用的手性辅基或配体。其中,具有第二重配位氮原子的手性亚砜配体,是最早应用于不对称过渡金属催化方法学研究的一类手性亚砜配体,并表现出较为优良的反应性能[1] (Scheme 1a)。然而,选择手性亚砜配体进行的合成转化研究,大多关注于采用钯催化剂促进的,通过各类对称亲电底物参与的不对称烯丙基烷基化方法学以及铑催化的苯硼酸与环烯酮之间的共轭加成反应方法学[1]-[2]。同时,由于现有的手性亚砜砌块,缺乏一定程度的多样性,进而阻碍上述砌块在有机合成设计中的进一步应用。并且,构建对映富集亚砜分子的传统策略主要涉及拆分技术、非对映选择性合成转化以及生物催化反应[3]。然而,对于涉及新型手性亚砜骨架构建的相关策略,则仍有待进一步研究。
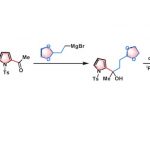
过渡金属催化的不对称C-H官能团化方法学是构建一系列复杂手性分子的重要策略之一。其中,对于选择高价过渡金属催化的C-H键活化方法学,最为成功的策略之一,则是采用廉价易得的外源手性酸 (external chiral acids) 作为配体,进而实现优良的立体选择性控制[4]。2008年,Yu课题组率先通过单重-N-保护的氨基酸 (mono-N-protected amino acid, MPAA)作为配体,成功完成首例通过Pd(II)催化剂参与的对映选择性C-H键活化反应方法学[4]-[5]。目前,MPAA已经成为Pd(II)催化的不对称C-H键活化方法学研究中,应用最为广泛的配体之一。并且,通过这一配体,能够有效地实现一系列具有点手性、平面手性以及轴手性的相关分子的构建[6]-[7]。同时,与构建具有碳立体中心手性分子的相关策略相比,通过不对称C-H活化策略进行的手性亚砜分子的构建,则较少有相关的文献报道。其中,Wang课题组报道通过Pd(II)催化的对映选择性C-H 烯基化策略,进而成功完成各类手性亚砜分子的构建[7],其反应过程涉及平行动力学拆分 (PKR)步骤以及采用Ac-Leu-OH作为配体,进行的去对称化步骤。之后,He课题组研究发现,通过Ir(III)催化的去对称化策略,同样能够有效地实现相应手性亚砜分子的构建,其反应过程同样涉及相关的PKR步骤 (Scheme 1b) [8]。值得注意的是,手性亚砜同样能够作为上述反应过程中的导向基团[8]。而且,通过上述的研究报道,同样表明,不对称C-H键活化策略,在一系列手性亚砜分子的合成中,具有良好的应用前景。
外消旋化合物的动力学拆分 (KR)是构建对映体纯有机分子的一种高效并实用的反应策略。而且,KR策略在不对称C-H活化中的应用研究,同样备受广泛关注[9]。近期,史炳锋课题组成功开发出一种通过Pd(II)催化剂促进的新型阻转选择性C-H活化策略,进而成功实现一系列轴手性苯乙烯与轴手性苯胺分子的构建[10]。其反应过程中采用廉价易得的L-焦谷氨酸 (L-pyroglutamic acid, L-pGlu-OH)作为手性配体。受到上述研究报道的启发,这里,本文成功设计出一种在Pd(II)催化剂与L-pGlu-OH手性配体存在下,通过2-(芳基亚磺酰基)吡啶底物参与的对映选择性C-H炔基化方法学,其反应过程中同样涉及KR步骤 (Scheme 1c)。
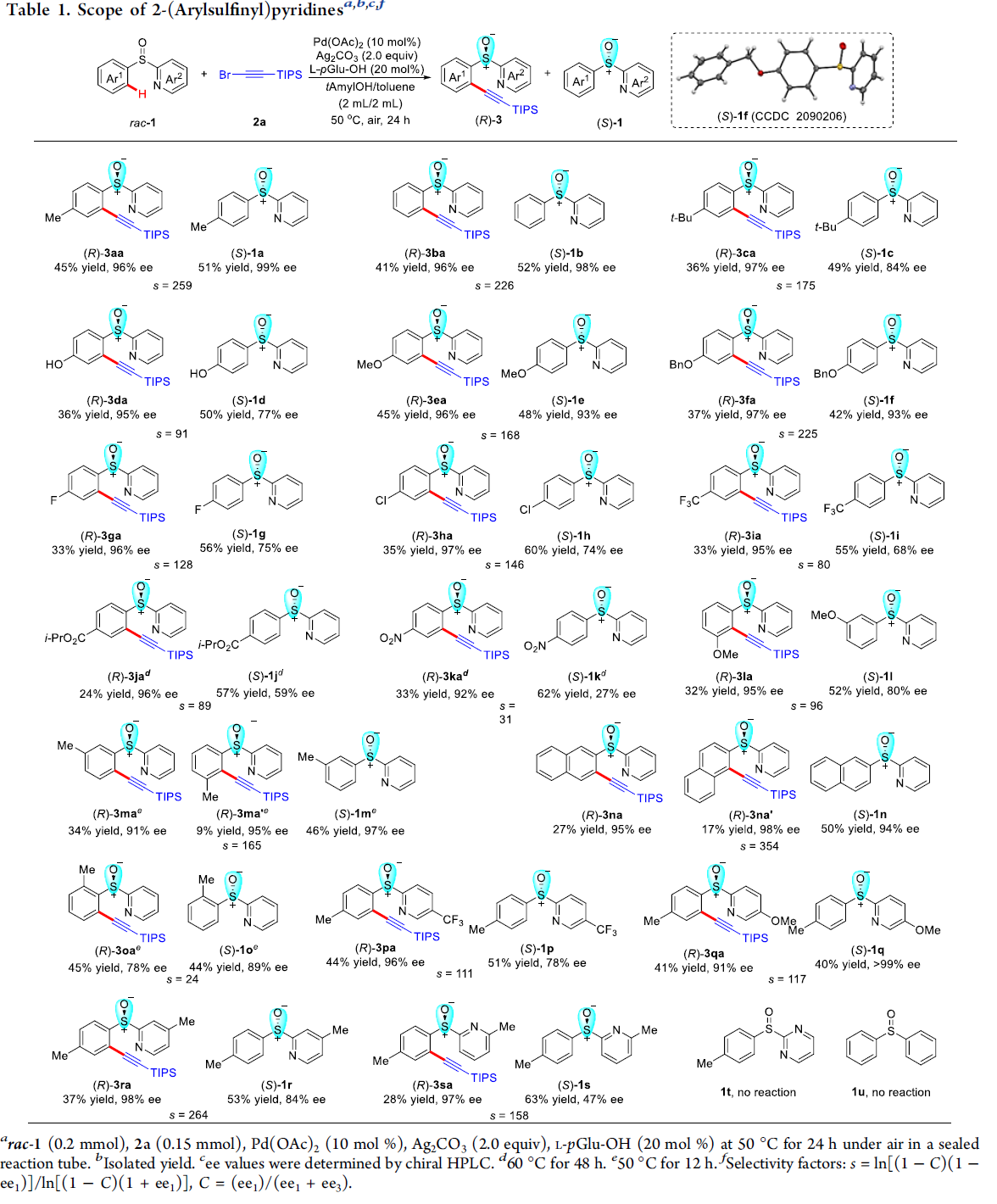
首先,作者采用rac–1a与2a作为模型底物,进行相关反应条件的优化筛选 (Table S1)。进而确定最佳的反应条件为:采用Pd(OAc)2作为催化剂,L-pGlu-OH作为手性配体,Ag2CO3作为银盐,在甲苯与tAmylOH的混合溶剂中,反应温度为50 oC,最终获得45%收率与96% ee的炔基化产物(R)-3aa以及51%收率与99% ee的非KR活性的手性亚砜起始原料 (S)-1a,其中,s值为259。

在上述的最佳反应条件下,作者首先对2-(芳基亚磺酰基)吡啶rac–1的底物应用范围进行考察 (Table 1)。研究表明,芳基对位中带有一系列供电子基团取代的2-(芳基亚磺酰基)吡啶底物 (1c–1f),在上述的标准反应条件下,均能顺利地与2a进行反应,并分别获得相应的手性产物(R)-3aa–3fa (36-45% 收率,95-97% ee)以及非KR活性的手性亚砜原料 (S)-1a–1f (42-52% 收率, 77-99% ee),并且,s值为91-259。同时,该小组发现,上述的标准反应条件,对于芳环中具有游离羟基取代的2-(芳基亚磺酰基)吡啶底物,同样能够良好地兼容。之后,研究发现,上述的最佳反应条件,对于芳基中具有吸电子基团取代的2-(芳基亚磺酰基)吡啶底物 (1g–1k),则表现出较低的反应活性,并获得相应的手性产物(R)-3ga–3ka (24-33% 收率,92-97% ee)以及非KR活性的手性亚砜原料 (S)-1g–1k (55-60% 收率,27-75% ee),s值为31-146。同时,该小组观察到,立体位阻对于上述的C-H炔基化过程无显著影响。在选择间位甲基取代的底物1m以及2-萘基取代的底物1n与2a进行相应的C-H炔基化反应时,能够同时获得具有较低以及较高立体位阻的邻位炔基化产物的混合物 (91-98% ee)。并且,该小组进一步发现,在选择间位甲氧基取代的底物1l时,相应的C-H炔基化过程仅能够在具有较高立体位阻的邻位进行,这可能源自于甲氧基与钯金属中心之间较弱的配位效应。接下来,作者观察到,在选择邻位甲基取代的底物1o时,由于立体效应的存在,最终无法获得具有高度对映选择性的炔基化产物(R)-3oa。此外,研究表明,对于吡啶环中具有不同取代基存在的2-(芳基亚磺酰基)吡啶底物 (1p–1s),同样能够有效地与上述的标准反应条件进行兼容。然而,上述的最佳反应条件对于底物1t与1u,则无法获得预期的炔基化产物。综上实验观察表明,上述的C-H炔基化过程中,吡啶作为导向基团。
接下来,作者对炔基溴底物的应用范围进行考察 (Table 2)。通过对上述反应条件进行进一步改进之后,作者观察到,具有较高立体位阻的炔丙基硅醚底物 2b以及具有较低立体位阻的炔基溴底物 2c与2d,均能够有效地与rac–1a顺利反应,并分别获得相应的手性产物(R)-3ab–3ad (11-41% 收率,90-97% ee)与非KR活性的手性亚砜原料 (S)-1a (42-79% 收率,21-89%ee)。

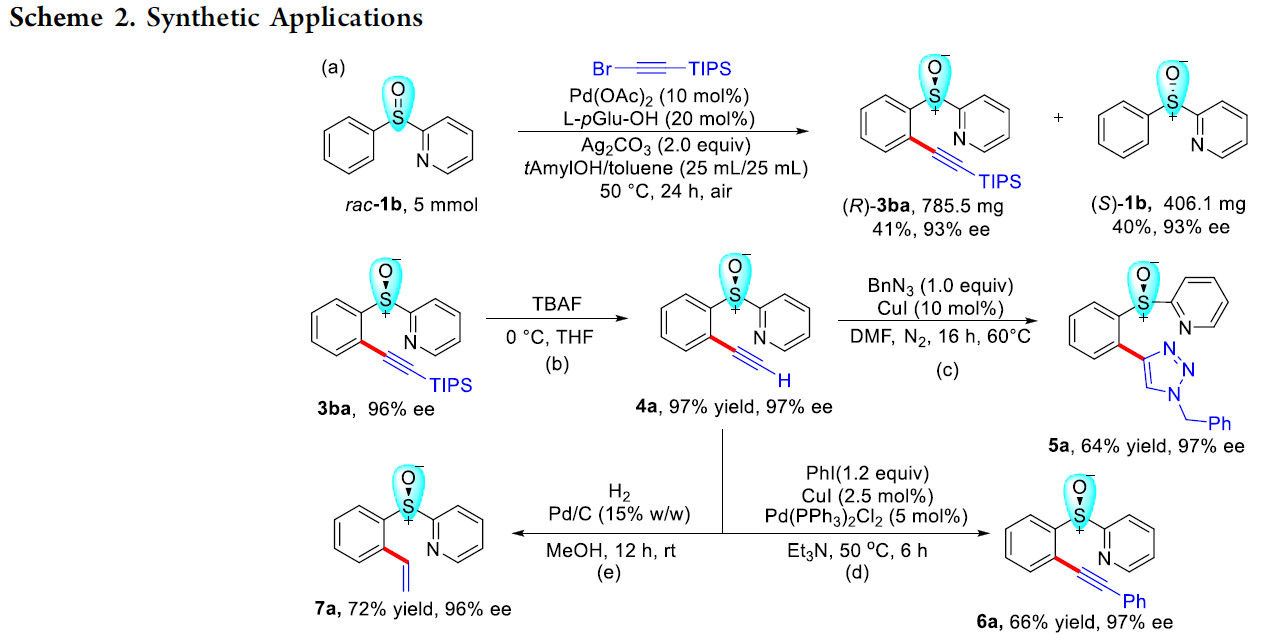
为进一步研究这一全新的C-H炔基化策略的合成实用性,接下来,作者对各类C-H炔基化产物进行相关的后期修饰研究 (Scheme 2)。首先,作者发现,将底物rac–1b的用量扩大至5 mmol时,同样能够获得41%收率与93% ee的手性炔基化产物 (R)-3ba以及40%收率与93% ee的非KR活性手性亚砜起始原料 (S)-1b (Scheme 2a)。同时,3ba中的TIPS基团,能够在温和的反应条件下,较为容易地去除,进而获得97%收率的端炔化合物4a (97% ee, Scheme 2b)。此外,端炔化合物4a能够分别通过click反应、sila-Sonogashira偶联反应以及还原反应过程,进一步获得化合物5a–7a (Scheme 2c, 2d与2e)。
总结
本文主要报道一种选择钯(II)催化剂促进的,通过2-(芳基亚磺酰基)吡啶底物参与的对映选择性C-H 炔基化反应方法学。反应过程中,选择廉价易得的L-pGlu-OH作为手性配体。同时,通过KR过程,能够以较高的反应收率与优良的对映选择性 (高达99%ee),获得相应的C-H炔基化产物。同时,作者进一步发现,通过上述过程形成的C-H炔基化产物,能够较为容易地转化为其它具有合成应用价值的重要砌块。
参考文献
[1] (a) J. V Allen, J. F Bower, J. M. J. Williams, Tetrahedron Asymmetry 1994, 5, 1895. doi: 10.1016/S0957-4166(00)86260-6.(b) J. K. Ekegren, P. Roth, K. Källström, T. Tarnai, P. G. Andersson, Org. Biomol. Chem. 2003, 1,358. doi: 10.1039/B208907F.
(c) H. Cheng, L. Lu, T. Wang, J. Chen, W. Xiao, Chem. Commun. 2012, 48, 5596. doi: 10.1039/C2CC31907A.
[2] (a) M. C. Carreno, G. Hernandez-Torres, M. Ribagorda, A. Urbano, Chem. Commun. 2009, 6129. doi: 10.1039/B908043K.(b) G. Sipos, E. E. Drinkel, R. Dorta, Chem. Soc. Rev. 2015, 44, 3834. doi: 10.1039/C4CS00524D.
(c) B. M. Trost, M. Rao, Angew. Chem. Int. Ed. 2015, 54, 5026. doi: 10.1002/anie.201411073.
[3] I. Fernandez, N. Khiar, Chem. Rev. 2003, 103, 3651. doi: 10.1021/cr990372u. [4] (a) K. M. Engle, J. Yu, J. Org. Chem. 2013, 78, 8927. doi: 10.1021/jo400159y.(b) Q. Shao, K. Wu, Z. Zhuang, S. Qian, J. Yu, Acc. Chem. Res. 2020, 53, 833. doi: 10.1021/acs.accounts.9b00621.
[5] B. Shi, N. Maugel, Y. Zhang, J. Yu, Angew. Chem. Int. Ed. 2008, 47, 4882. doi: 10.1002/anie.200801030. [6] (a) B. Shi, Y. Zhang, J. K. Lam, D. Wang, J. Yu, J. Am. Chem. Soc. 2010, 132, 460. doi: 10.1021/ja909571z.(b) X. Cheng, Y. Li, Y. Su, F. Yin, J. Wang, J. Sheng, H. U. Vora, X. Wang, J. Yu, J. Am. Chem. Soc. 2013, 135, 1236. doi: 10.1021/ja311259x.
(c) L. Chu, K. Xiao, J. Yu, Science 2014, 346, 451. doi: 10.1126/science.1258538.
(d) L. Hu, P. Shen, Q. Shao, K. Hong, J. Qiao, J. Yu, Angew. Chem. Int. Ed. 2019, 58, 2134. doi: 10.1002/anie.201813055.
(e) D. Gao, Y. Shi, Q. Gu, Z. Zhao, S. You, J. Am. Chem. Soc. 2013, 135, 86. doi: 10.1021/ja311082u.
(f) C. Pi, Y. Li, X. Cui, H. Zhang, Y. Han, Y. Wu, Chem. Sci. 2013, 4, 2675. doi: 10.1039/C3SC50577D.
[7] Y. Zhu, Y. Li, B. Zhang, F. Zhang, Y. Yang, X. Wang, Angew. Chem. Int. Ed. 2018, 57, 5129. doi: 10.1002/anie.201801146. [8] W. Liu, W. Yang, J. Zhu, Y. Guo, N. Wang, J. Ke, P. Yu, C. He, ACS Catal. 2020, 10, 7207. doi: 10.1021/acscatal.0c02109. [9] (a) M. Brauns, N. Cramer, Angew. Chem. Int. Ed. 2019, 58, 8902. doi: 10.1002/anie.201904543.(b) Y. Sun, N. Cramer, Chem. Sci. 2018, 9, 2981. doi: 10.1039/C7SC05411D.
(c) Q. Yao, S. Zhang, B. Zhan, B. Shi, Angew. Chem. Int. Ed. 2017, 56, 6617. doi: 10.1002/anie.201701849.
(d) K. Xiao, L. Chu, G. Chen, J. Yu, J. Am. Chem. Soc. 2016, 138, 7796. doi: 10.1021/jacs.6b04660.
(e) K. Xiao, L. Chu, J. Yu, Angew. Chem. Int. Ed. 2016, 55, 2856. doi: 10.1002/anie.201510808.
(f) D. Gao, Q. Gu, S. You, ACS Catal. 2014, 4, 2741. doi: 10.1021/cs500813z.
[10] (a) L. Jin, Q. Yao, P. Xie, Y. Li, B. Zhan, Y. Han, X. Hong, B. Shi, Chem. 2020, 6, 497. doi: 10.1016/j.chempr.2019.12.011.(b) Q. Yao, P. Xie, Y. Wu, Y. Feng, M. Teng, X. Hong, B. Shi, J. Am. Chem. Soc. 2020, 142, 18266. doi: 10.1021/jacs.0c09400.
(c) Y. Wu, P. Xie, G. Zhou, Q. Yao, X. Hong, B. Shi, Chem. Sci. 2021, 12, 9391. doi: 10.1039/D1SC01130H.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载















No comments yet.