
本文作者:杉杉
导读
近日,California大学的N. K. Garg教授课题组在JACS中发表论文,报道一种在钯催化条件下,通过原位形成的张力环联烯 (strained cyclic allene) 参与的环化反应方法学。这一全新的环化策略中采用芳卤与环联烯前体,作为反应参与物,进而成功完成一系列稠杂环分子的构建。同时,上述的环化过程中涉及两种不同的新键以及一个sp3中心的形成。此外,这一新型的环化策略同样能够良好地实现非对映选择性以及对映选择性的控制,进而获得一系列结构复杂的手性分子。
Palladium-Catalyzed Annulations of Strained Cyclic Allenes
A. V. Kelleghan, D. C. Witkowski, M. S. McVeigh, N. K. Garg, J. Am. Chem. Soc. 2021, 143, 9338. doi: 10.1021/jacs.1c04896.
正文
自20世纪初以来,瞬态张力环中间体 (transient strained cyclic intermediate) 例如苯炔 (1, Figure 1A),一直备受化学家的广泛关注。20世纪初,在提出苯炔作为反应活性中间体的假设之后,这一中间体的存在,一直以来备受学术界的争议。然而,直到20世纪50年代,通过Roberts[1]与Wittig[2]-[3]的开创性实验研究,进而证实这一中间体的存在。由于苯炔中间体中存在高度的环张力,因而,能够表现出较为独特的反应活性。并且,通过苯炔中间体相关研究的启发,目前,已经成功设计出一系列相关的杂环芳炔中间体,例如2与3,以及饱和环炔类似物,例如环己炔 4。同时,上述的张力环炔中间体在当代合成工具箱 (modern synthetic toolbox)中发挥极为重要的作用,目前,已经广泛应用于杂环、催化配体、天然产物、农用化学品以及有机材料分子的构建。
尽管张力环炔中间体的研究,目前已经受到合成化学家的广泛关注。然而,对于具有较高环张力的瞬态环联烯中间体 (例如1,2-环己二烯5) 的研究,则较少有相关的文献报道[4]。值得注意的是,张力环联烯与张力环炔中间体具有较为相近的环张力与反应活性[5]。并且,通过环联烯中间体,能够顺利实现一系列富含sp3立体中心的手性分子以及立体化学更为复杂的相关有机分子的构建[6]。然而,迄今为止,上述中间体相关的方法学研究主要涉及三种不同类型的分子间反应过程 (Figure 1B),即亲核捕获 (例如7+KOt-Bu→6)、环加成 (例如7+8→9)以及采用过渡金属催化的环联烯捕获反应 (例如7+10→11)[7]-[8]。然而,由于反应过程中,需要使两种较低浓度的活性中间体,即催化产生的中间体与原位形成的瞬态环联烯中间体,之间进行有效的反应,因此,这一策略具有其内在的动力学挑战性。
目前,仅有两例涉及过渡金属催化的,并通过张力环联烯中间体进行的合成转化方法学的相关研究报道,即2009年,Guitián等[7]报道的首例Pd催化的环联烯[2+2+2]反应方法学以及在2020年,本课题组[8]报道的采用Ni催化剂促进的环联烯与苯并三嗪 (benzotriazinones)之间的环化反应方法学。并且,通过上述研究表明,选择过渡金属催化与环联烯的结合,能够为张力中间体化学 (strained intermediate chemistry)的研究开辟出一条全新的途径。

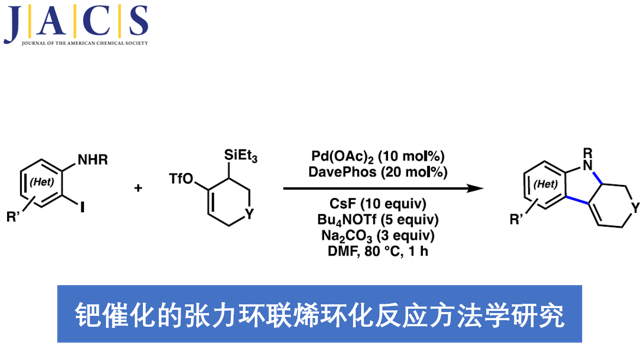
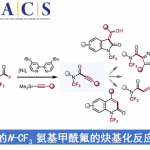
受到上述研究报道的启发,作者设计出一种全新的通过张力环联烯参与的环化反应策略 (Scheme 1)。作者设想,首先,通过具有芳基碘以及亲核性XH结构单元的双功能环化参与物12与过渡金属催化剂之间的氧化加成过程,形成中间体15。同时,硅基三氟甲磺酸酯13通过氟离子促进的去硅基化过程,原位形成张力环联烯中间体7。之后,通过环联烯中间体7与15中Ar-M键的迁移插入过程,形成η3-配合物16。接下来,通过配合物16进行后续的亲核环化与还原消除过程,即可获得相应的三环产物14。基于上述的研究设想,这里,本课题组成功开发出一种在钯催化剂存在的条件下,通过原位形成的张力环联烯中间体参与的环化反应方法学。并通过这一全新的环化反应策略,成功实现一系列稠合多环分子的构建。同时,作者进一步发现,这一全新的环化策略同样能够有效地实现优良的立体选择性与对映选择性的控制。
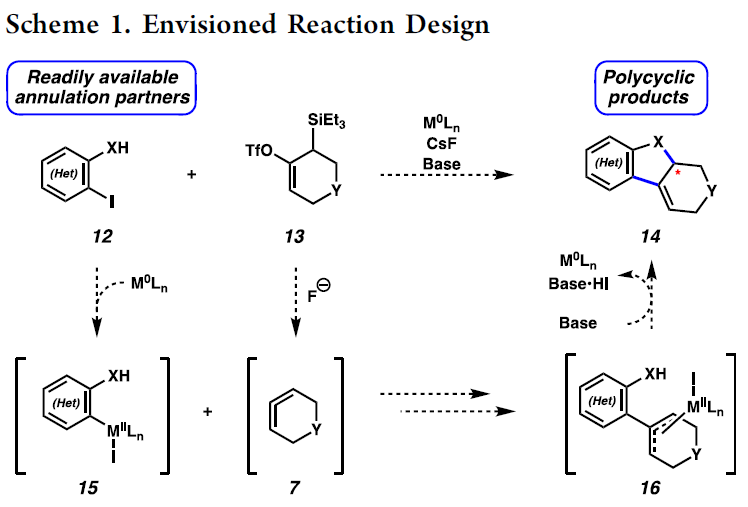
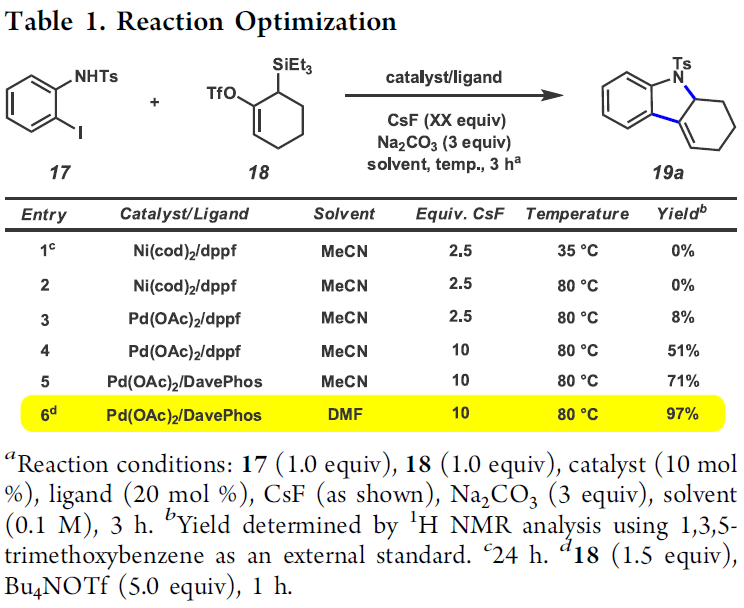
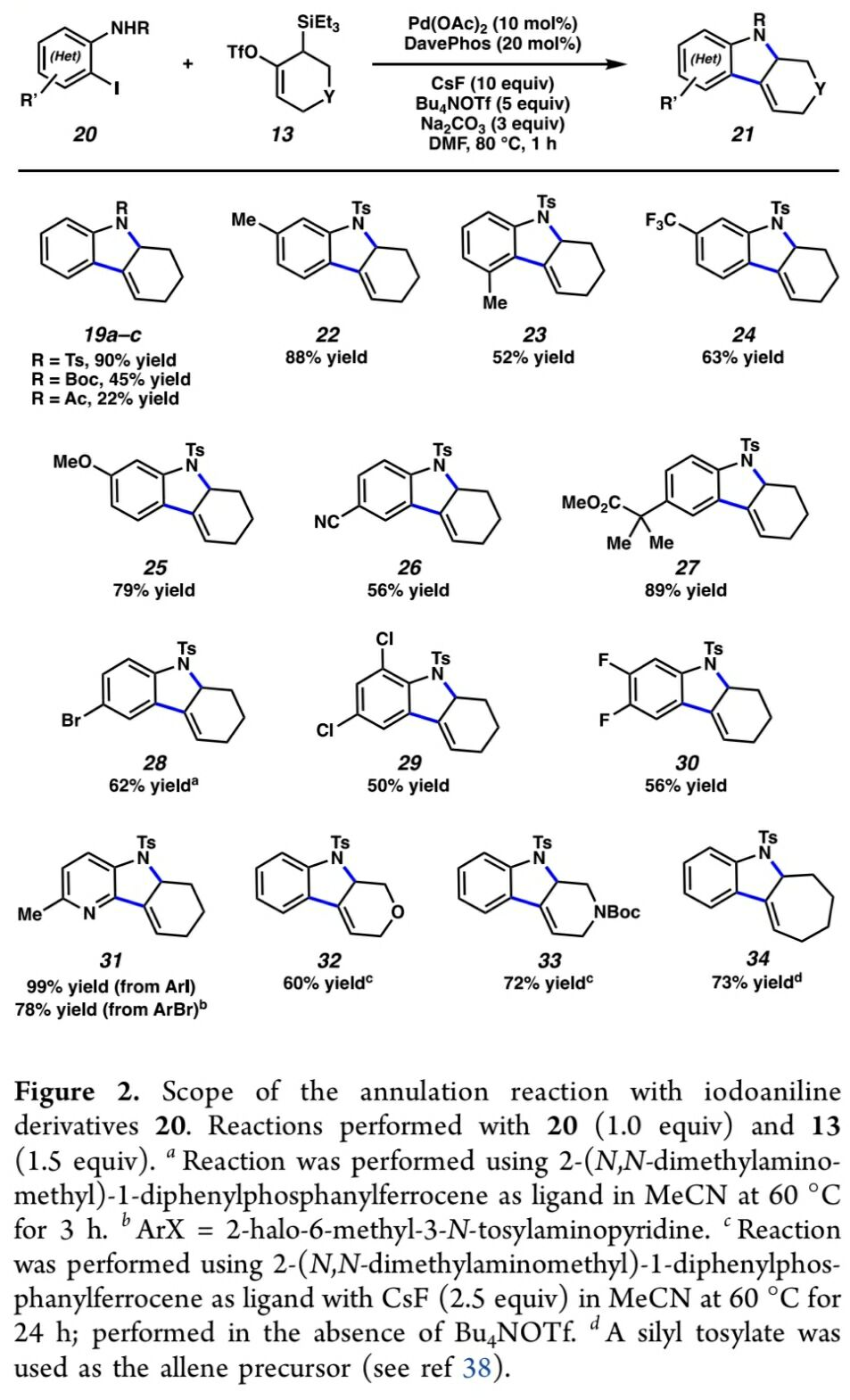
首先,作者采用邻碘苯胺衍生物17与硅基三氟甲磺酸酯衍生物18作为模型底物,进行相关环化反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用Pd(OAc)2作为催化剂,DavePhos作为配体,DMF作为反应溶剂,Bu4NOTf与CsF作为添加剂以及Na2CO3作为碱,反应温度为80 oC,最终获得97%收率的目标产物19a。
在获得上述的最佳反应条件之后,作者开始对反应过程中的底物应用范围进行研究 (Figure 2)。首先,该小组发现,邻碘苯胺衍生物中的N–取代基为-Ts、-Boc以及-Ac时,均能够顺利地参与上述的环化过程,并获得相应的环化产物19a–19c。同时,上述的标准反应条件,对于邻碘苯胺底物苯基中存在的一系列电子与立体效应不同的取代基团,均能够良好地兼容,并获得相应的四氢咔唑产物22–30。同时,反应过程中表现出优良的官能团兼容性。之后,该小组进一步对各类杂环底物以及杂环联烯前体的应用范围进行深入研究。实验表明,具有溴吡啶基以及碘吡啶基的杂环底物同样能够经历预期的环化过程,并以良好至优良的反应收率获得相应目标产物31。接下来,作者发现,作为环联烯前体的硅基三氟甲磺酸酯13的相应氧杂与氮杂衍生物,均能够较好地与上述的标准反应体系兼容,并获得相应的目标产物32–33。并且,作者进一步观察到,七元环联烯前体同样能够顺利地参与上述的环化过程,并获得相应的杂环产物34。
之后,作者对进一步其它不同类型的前亲核底物进行深入研究 (Figure 3)。实验表明,O-前亲核底物 35与37在上述的最佳反应条件下,分别能够获得相应的环化产物36 (87%反应收率)与38 (46%反应收率)。之后,作者发现,选择Xantphos作为配体时,在上述的最佳反应条件下,采用N-前亲核底物 (39),同样能够获得71%收率的目标产物40。接下来,该小组观察到,碳基前亲核底物(C-base pronucleophile) 41在上述的标准反应条件下,同样能够获得60%收率的具有四级碳中心的四氢芴分子42。

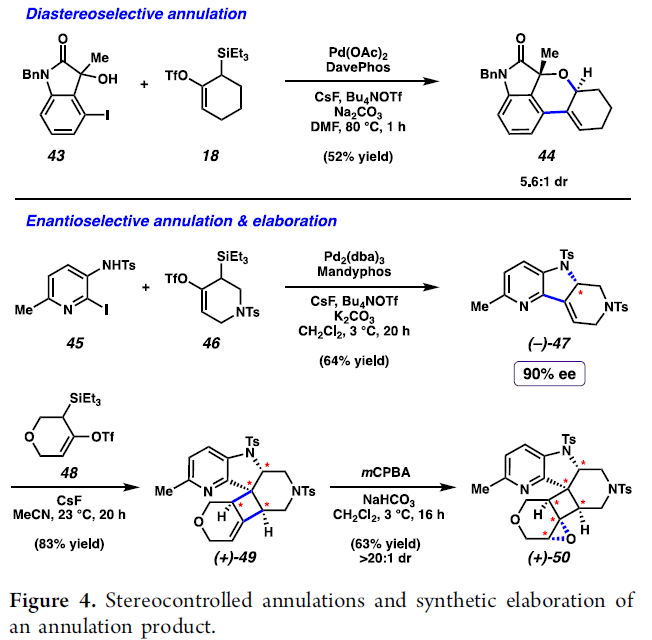
此外,作者同样对上述环化过程中的非对映与对映选择性的控制,进行深入考察 (Figure 4)。首先,作者发现,三级苄醇43能够与联烯前体18在上述的标准环化条件下进行反应,并获得相应的四环产物44 (5.6:1 d.r.)。之后,该小组选择碘吡啶45与联烯前体46在Pd2(dba)3/Mandyphos催化剂体系中进行反应,最终获得90% ee的三环产物 (-)-47。并且,该小组发现,三环产物(-)-47能够继续与氧杂联烯前体 (oxacyclic allene precursor)48进行反应,进而获得相应产物(+)-49。之后,(+)-49中的烯基结构单元进一步经历立体选择性性的环氧化过程,最终获得对映富集的六环分子 (+)-50。
总结
California大学的N. K. Garg教授课题组报道一种在钯催化条件下,通过原位形成的张力环联烯中间体进行的环化反应方法学,其中采用芳卤与环联烯前体作为反应底物,进而成功完成一系列稠杂环分子的构建。同时,作者进一步对上述环化策略中非对映选择性与对映选择性的控制进行深入研究,并获得相应的产物44与(-)-47。此外,作者发现,通过(-)-47作为关键砌块,能够进一步获得对映富集的六环分子 (+)-50。
参考文献
[1] J. D. Roberts, H. E. Simmons, L. A. Carlsmith, C. W. Vaughan, J. Am. Chem. Soc. 1953, 75, 3290. doi: 10.1021/ja01109a523. [2] G. Wittig, Naturwissenschaften 1942, 30, 696. doi: 10.1007/BF01489519. [3] G. Wittig, L. Pohmer, Angew. Chem. 1955, 67, 348. doi: 10.1002/ange.19550671306. [4] M. Christl, Cyclic allenes up to seven-membered rings. Modern Allene Chemistry, N. Krause, S. A. K. Kashmi Eds. Wiley-VCH: Weinheim, 2004, 243. [5] G. Wittig, P. Fritze, Angew. Chem. Int. Ed. 1966, 5, 846. doi: 10.1002/anie.196608461. [6] F. Lovering, J. Bikker, C. Humblet, J. Med. Chem. 2009, 52, 6752. doi: 10.1021/jm901241e. [7] I. Quintana, D. Peña, D. Pérez, E. Guitián, Eur. J. Org. Chem. 2009, 5519. doi: 10.1002/ejoc.200900631. [8] M. M. Yamano, A. V. Kelleghan, Q. Shao, M. Giroud, B. J. Simmons, B. Li, S. Chen, K. N. Houk, N. K. Garg, Nature 2020, 586, 242. doi: 10.1038/s41586-020-2701-2.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!















No comments yet.