
本文作者:ChemBoy
导读
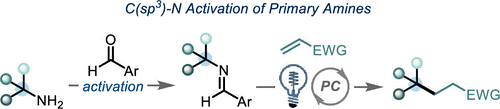
伯胺是一种廉价、天然存在且化学多样性的化学原料,因而关于胺的脱胺基官能团化已经成为一个重要的研究方向。近期,随着Katritzky吡啶盐类试剂的出现,已实现了简单α-1o以及α-2o胺的C-N键活化转化。最近,美国哥伦比亚大学Tomislav Rovis研究团队通过可见光光氧化还原催化成功实现了具有位阻的α-3o伯胺的脱氨基烷基化反应,涉及α-3o伯胺与富电子芳醛发生缩合,再经氧化脱质子化过程生成关键中间体亚胺自由基,随后发生β-切断释放出烷基从而实现自由基实现α-3o伯胺C-N键切断的过程。相关研究成果于近期发表在《J. Am. Chem. Soc.》上:
“Photoredox-Catalyzed Deaminative Alkylation via C−N Bond Activation of Primary Amines”
Melissa A. Ashley and Tomislav Rovis*
J. Am. Chem. Soc. 2020, 142, 18310. DOI: 10.1021/jacs.0c08595 https://doi.org/10.1021/jacs.0c08595
正文
伯胺广泛存在于各种合成砌块中、并且在有机合成中用途广泛。有机化学家报道了很多胺的N-H键官能团化,如还原胺化[1]或交叉偶联反应[2]。此外,利用胺作为C-H键活化的导向基团可实现位点选择性的C-H键官能团化[3]。尽管已经取得了一定的研究进展,但是利用胺的C-N键断裂进行脱胺化转化仍未被充分利用。目前,C-N键的功能化仅局限于烯丙基或苄基胺(Scheme 1A)[4]。最近,随着Katritzky盐的出现,利用一级胺作为烷基前体引起了有机合成化学家的广泛关注,并已经被用于各种过渡金属催化和光氧化还原催化的脱氨基官能团化反应[5]。广受欢迎的Katritzky盐实现了无催化剂的C-N键活化过程,然而这种活化方法仅适用于位阻较小的α-1o以及α-2o胺,而位阻较大的α-3o胺并不适用。
作者在这里有两个目标:1)发展出一种活化大位阻一级胺的C-N键活化方法;2)通过氧化过程释放出烷基自由基,为Katritzky盐活化方法提供一种补充。芳香醛是形成亚胺自由基的一种潜在活化官能团,亚胺自由基可能发生β-断裂释放出烷基自由基[6]。在以前的报道中,亚胺自由基是通过自由基与异氰酸酯发生加成产生。而作者受已经报道的利用质子偶合电子转移的策略对C-H键进行活化反应的启发,假设富电子的亚胺经过单电子氧化与去质子化也能生成亚胺基自由基。亚胺通常是通过单电子还原过程被活化。然而,作者设想是否可以通过芳烃发生单电子氧化,从而促进亚胺基C-H键的去质子化。如Scheme 1C所示,甲苯(Ep/2 = 2.36 V vs SCE)被氧化形成的自由基阳离子具有格外强的π-酸性((pKa of -6)。作者想将这种策略应用于亚胺的活化生成亚胺基自由基的过程中。
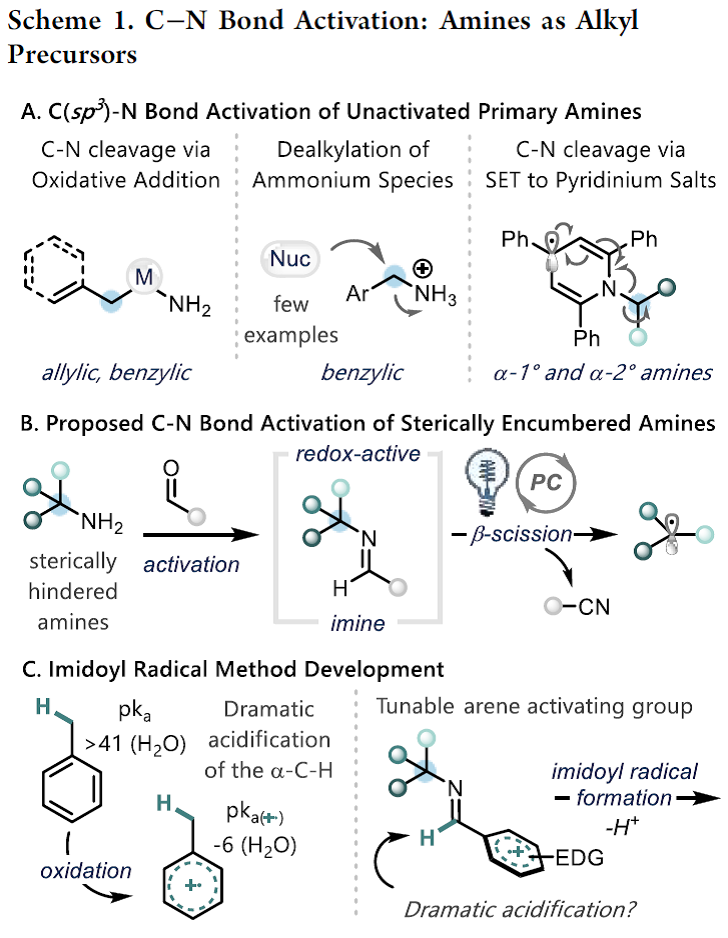
有了以上的设想,接下来作者就对各种醛活化的金刚烷基亚胺的氧化电势进行研究,发现2,4,6-三甲氧基苯甲醛是一种可行的活化基团。作者就以这种醛与金刚烷基胺形成的亚胺1作为模板底物进行反应条件筛选(Table 1)。首先,作者发现在光催化PC A、三氟甲苯作为溶剂、磷酸钾的存在条件下,亚胺1与丙烯酸酯2以19 %的产率生成目标产物3(Table 1, entry 1)。随后作者继续对光催化剂、碱、反应溶剂以及反应浓度等反应参数进行筛选后发现[Ir(dF-CF3-ppy)2(dtbbpy)PF6](PC B, Ir(III)*/Ir(II) = 1.21 V vs SCE; 3 mol%)为光催化剂、1,1,3,3-四甲基胍(TMG, 20 mol%)作为碱、1,2-二氯乙烷(DCE, 0.05M)作为反应溶剂、在波长为427 nm的蓝光辐射下,在室温条件下金刚烷基亚胺1与烯烃4以91%的产率生成目标产物γ-季碳中心氨基酸(Table 1, entry 11),作者以此反应条件作为最优反应条件。
在确定最优反应条件后,作者开始对反应的底物普适性进行研究(Scheme 2)。首先,作者对α-3o伯胺底物范围进行了探索。金刚烷基胺衍生类底物能够分别以92%(5)与75%(6)的产率生成目标化合物。作者发现无论非环状还是环状α-3o胺均能以较高的产率(57-99 %)生成目标产物(7-18),其中小分子药物phenantramine(10,99%)及其氟化衍生物(11, 83%)也能表现出很高的反应活性。此外,作者还对一系列的缺电子烯烃偶联体进行了探索。如简单丙烯酸酯、α-取代的丙烯酸酯类衍生物、乙烯基砜以及甲基丙烯腈均能在该反应体系中顺利进行反应,以中等至优秀的产率(35-87%)生成目标产物(19-30)。作者发现,α-取代的丙烯酸酯类烯烃偶联体能够有效提高反应的产率(22-26),作者认为这是因为α-取代基能够有效抑制3o烷基自由基与烯烃加成生成的自由基中体的自身聚合反应已经其他官能团转移反应的发生。

接下来,作者对该反应的机理进行了研究探索以确定C-N键活化与烷基自由基形成的模式(Scheme 3)。通过原位LED NMR监测结果表明,亚胺与丙烯酸酯偶联体的反应是一级动力学反应(Scheme 3B)。基于这样的结果,作者推测一级胺与醛活化基团缩合生成的氧化还原活性亚胺能成功猝灭激发态的光催化剂PC B,通过Stern-Volmer荧光猝灭实验证明了这一推测(Scheme 3C)。接着,经质子转移形成的亚氨基自由基中间体,随后发生β-断裂释放出烷基自由基,该烷基自由基与缺电子烯烃发生加成形成新的烷基自由基中间体,该新烷基自由基中间体被二价光催化剂还原并质子化生成目标产物,同时基态光催化剂PC B再生。作者还对亚胺基自由基的形成过程进行了研究,他们认为亚胺自由基的形成有两种可能的路径: 1)N-自由基阳离子作为氢原子转移试剂(HAT)或者2)芳烃被氧化接着形成的中间体酸性亚氨基氢发生去质子化生成亚胺基自由基。为了证明氢原子转移路径,作者在34与35间做了竞争实验(Scheme 3E),通过实验结果,作者否定了氢原子转移生成亚氨基自由基中间体的路径。从而作者认为底物被氧化的发生时C-N键活化发生的关键。此外,作者通过理论计算测定了亚胺基C-H键的解离能,以及通过实验测定了其氧化电势(Ep/2 = 1.43 V vs SCE),作者估算出亚胺基阳离子的pKa约为15。这种被高度酸化的亚氨基C-H键很容易发生去质子化。接着自旋中心的移位导致亚胺自由基的形成,随着发生β-断裂释放出烷基自由基,紧接着与缺电子烯烃发生偶联(Scheme 3F)。

总结
Rovis课题组成功发展了一种可见光氧化还原催化的α-3o伯胺的脱氨基烷基化反应方法。该方法通过形成氧化还原活性的亚胺活化大位阻胺的C-N键,该亚胺经氧化后导致亚胺基C-H键酸性明显增强,经去质子化和自旋中心位移形成了关键的亚胺自由基中间体。该中间体经β-断裂生成亲核性的烷基自由基,其与缺电子烯烃偶联得到非天然的γ-季碳氨基酸衍生物。同时,该方法是Katritzky盐活化C-N键方法的一种补充。
(注:本文中所有图片均来自J. Am. Chem. Soc. 2020, 142, 18310)
参考文献
[1] Afanasyev, O. I.; Kuchuk, E.; Usanov, D. L.; Chusov, D. Chem. Rev. 2019, 119, 11857. DOI:. 10.1021/acs.chemrev.9b00383.[2] Ruiz Castillo, P.; Buchwald, S. L. Chem. Rev. 2016, 116, 12564. DOI: 10.1021/acs.chemrev.6b00512.
[3] Beatty, J. W.; Stephenson, C. R. J. Acc. Chem. Res. 2015, 48, 1474. DOI: 10.1021/acs.accounts.5b00068.
[4] Wang, O.; Su, Y.; Li, L.; Huang, H. Chem. Soc. Rev. 2016, 45, 1257.DOI: 10.1039/c5cs00534e.
[5] Correia, J. T. M.; Fernandes, V. A.; Matsuo, B. T.; Delgado, J. A. C.; de Souza, W. C.; Paixao, M. W. Chem. Commun. 2020, 56, 503.DOI:10.1039/c9cc08348k.
[6] Dauncey, E. M.; Morcillo, S. P.; Douglas, J. J.; Sheikh, N. S.; Leonori, D. Angew. Chem., Int. Ed. 2018, 57, 744. DOI: 10.1002/anie.201710790.















No comments yet.