
本文作者:杉杉
导读
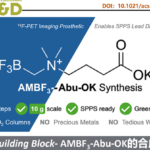
诺华生物医学研究所(Novartis Institutes for BioMedical Research)Markus Furegati研究团队报道了一种新型β-内酰胺酶抑制剂IID572合成的新途径,克服了文献中总收率低(0.9%)且关键起始底物不易获得的弊端。通过此方案,可实现从市售的底物开始,并以19步获得1.7-2.9%总产率的目标化合物IID572。该方案主要通过亚胺叶立德[3+2]环加成反应制备复杂的中间体,同时通过酶拆分方法获得所需的对映异构体(高ee值)。尽管在官能团相互转化方面存在一些合成挑战,但仍可以进行8 mol规模的放大,从而获得10 g的IID572以及各类中间体,为临床前毒理学研究和各类反应条件的优化提供了便捷。
Scale up synthesis of IID572: A new-lactamase inhibitor
Markus Furegati, Sandro Nocito, Folkert Reck, Anthony Casarez, Robert Simmons, Heiner Schuetz, and Guido Koch
Org. Process Res. Dev. ASAP DOI: 10.1021/acs.oprd.0c00069

正文
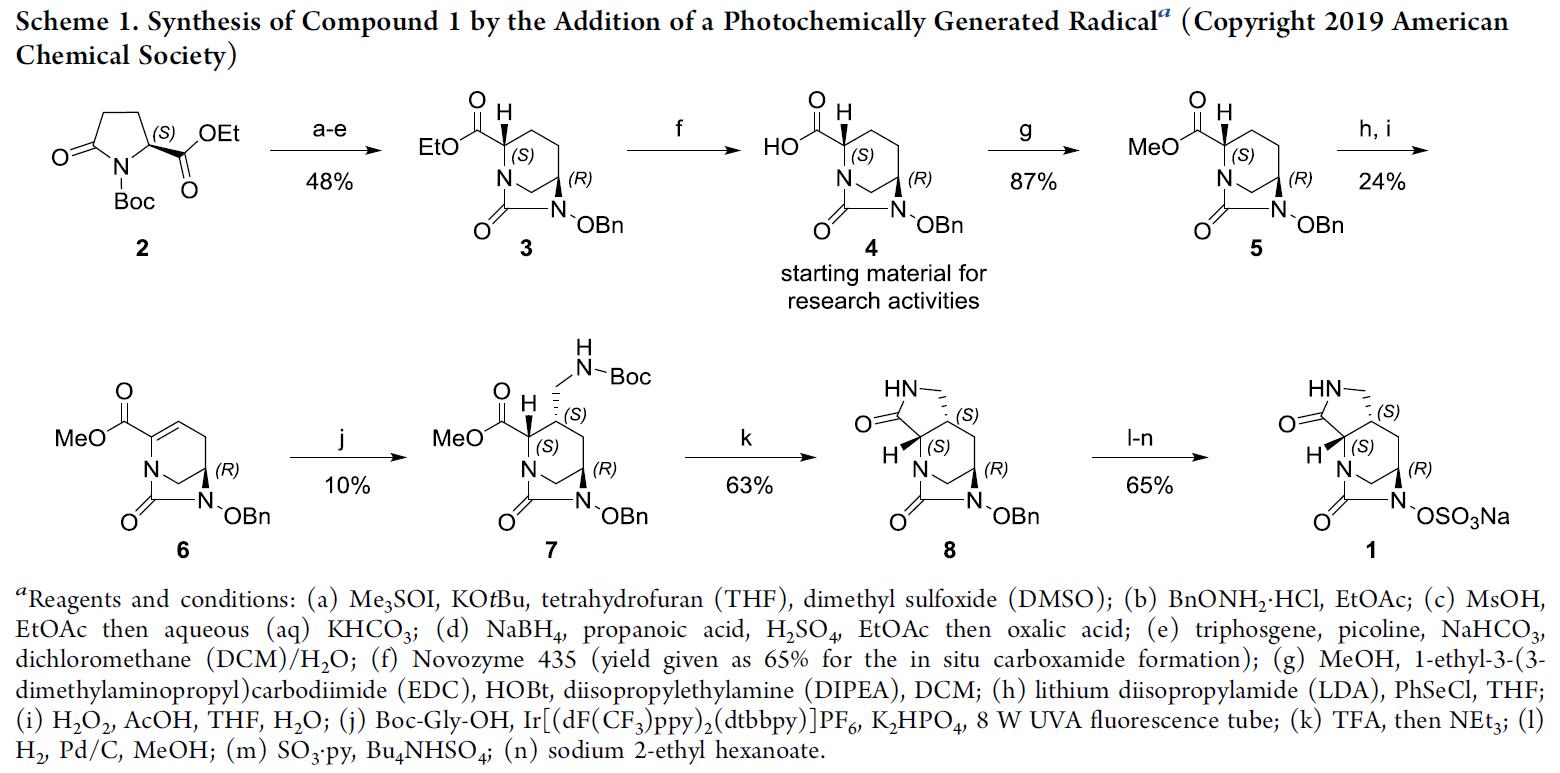
国内外用于临床治疗的β-内酰胺酶抑制剂合剂种类很多,如阿莫西林、替卡西林、氨苄西林、舒拉西林、头孢哌酮等。临床应用显示,这些合剂对某些β-内酰胺酶所致的耐药菌株具有明显抑制作用,但对不同种类β-内酰胺酶的抑制作用范围有限。因此,新型的β-内酰胺酶抑制剂仍在不断地开发。在对二氮杂双环辛烷(DBO)类新型β-内酰胺酶抑制剂的研究中,作者发现,IID572(1)作为最佳的抑制剂,可与LYS228或其他β-内酰胺类抗生素联合使用,对抗由更广泛的CRE引起的难以治疗的感染。而后期功能化(LSF)则是合成IID572的有效方法。据文献报道,该路线的起点是阿维巴坦(Avibactam)合成的中间体4(Scheme 1),可在6个步骤中以48%的收率获得。该方案的关键步骤为在光化学下Michael受体6进行Giese自由基加成反应,从而获得中间体7和三种其他非对映异构体的混合物。分离出所需的异构体7,再用TFA脱保护后,伯胺在碱性条件下自发环化,得到化合物8。
虽然该工艺路线相对较短,也对于LSF来说比较理想,但在按比例放大方面存在三个弊端。首先,需要PhSeCl(一种有毒且昂贵的试剂)来引入自由基受体烯烃,仅以24%的收率获得6。其次,在自由基加成之后形成非对映异构混合物,仅形成10%的所需异构体7。四种异构体的比例不受反应条件的影响(如溶剂、催化剂或温度)。第三,即使从乙酯3开始经12步仅以0.48%的总产率的获得目标产物1。
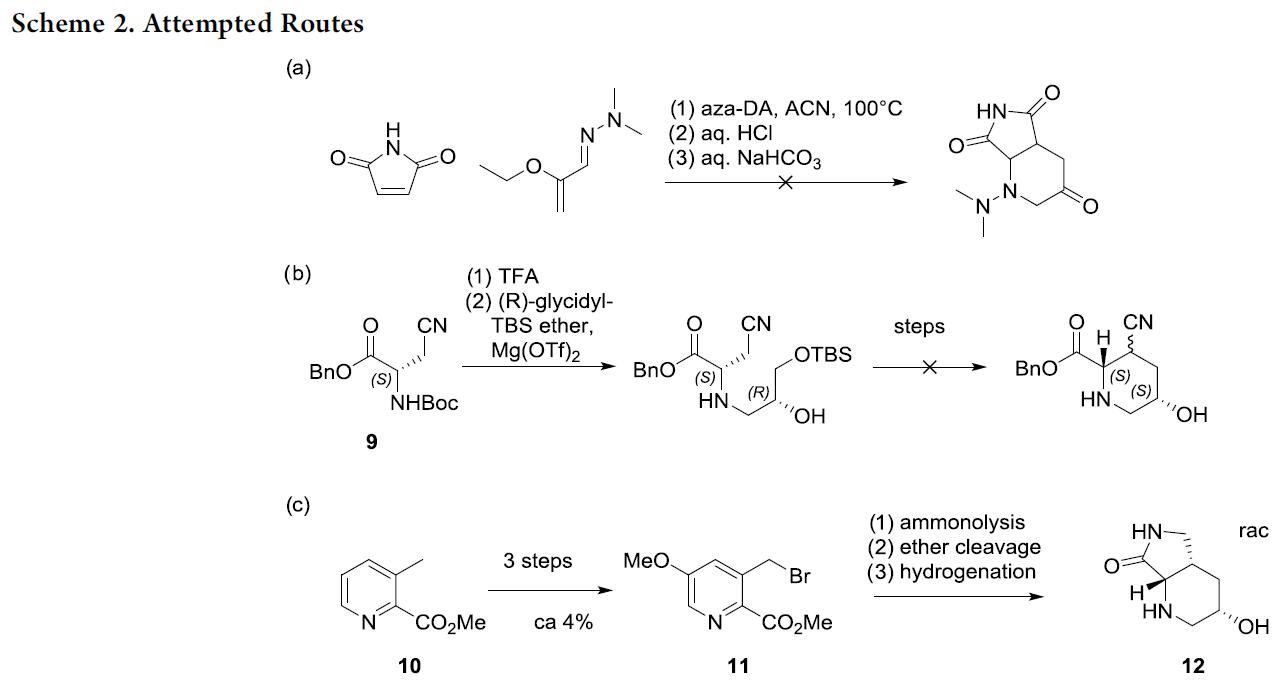
随后,作者进行了三种路线的设计(Scheme 2)。(a)Aza-Diels-Alder路线,然而,环加成作为具有挑战性的关键步骤,但结果获得复杂的混合物。(b)天冬酰胺环化路线,以氨基酸衍生的9为底物,然而N-烷基化效果不好,同时环化失败。(c)从已知的11中进行吡啶加氢,尽管这是一种有前途的方法,但以昂贵的3-甲基吡啶-2-羧酸甲酯(10)开始,同时不能获得令人满意的收率,氨解也不能很好地进行。因此,上述三种路线均被放弃。
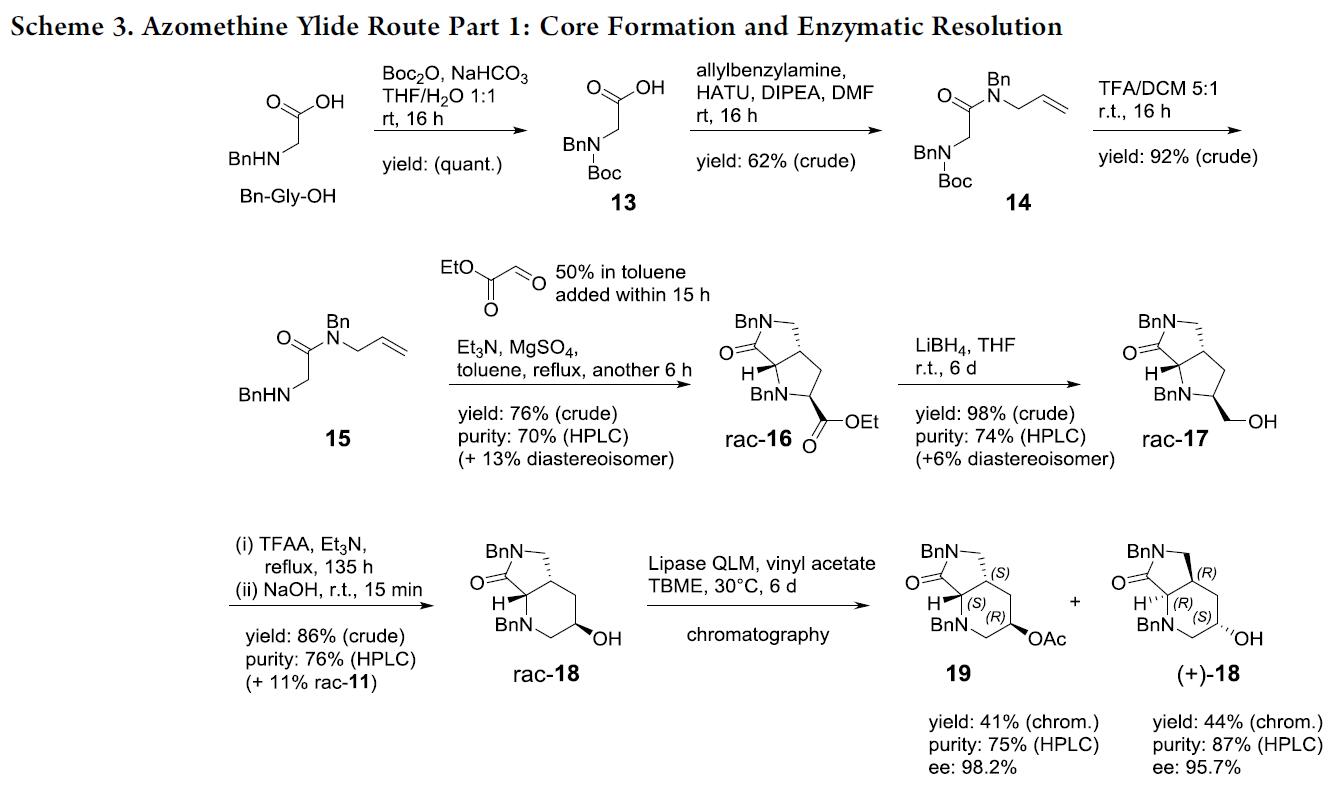
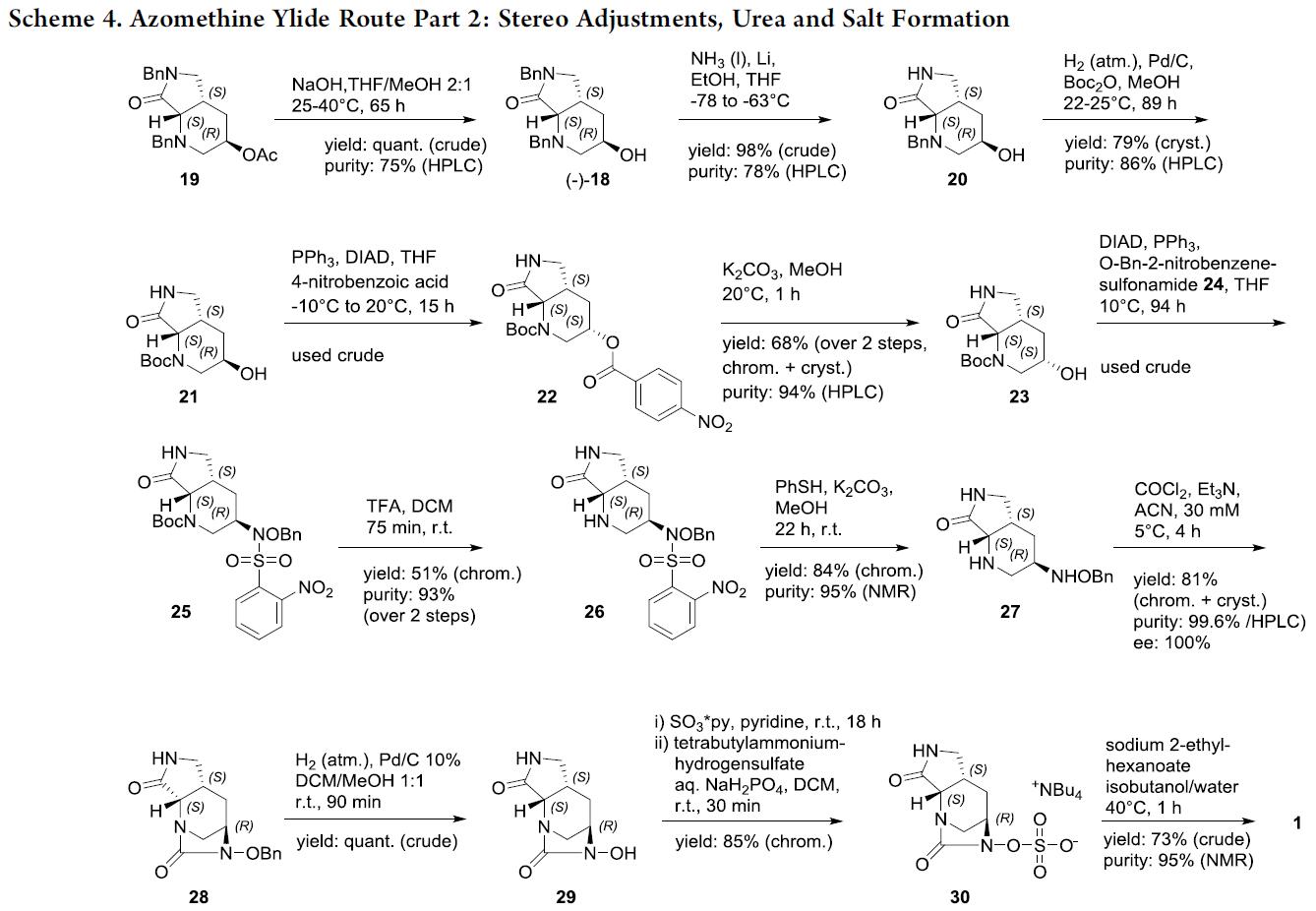
因此,作者选择简单易得的起始原料,并通过分子内甲亚胺叶立德[3+2]环加成反应以构建复杂分子结构(Scheme 3)。根据文献中报道,当在NaHCO3存在下,可在水/THF中以定量收率获得Bn-Gly-OH的Boc保护中间体13。同时,T3P被证明是与市售烯丙基苄胺形成酰胺的最佳偶联剂,并通过TFA脱Boc保护获得N-烯丙基-2-(甲基氨基)-N-苯基乙酰胺15,收率为30%;若缓慢滴加乙醛酸乙酯时,可将收率提高至76%。同时,该反应主要副产物是非对映异构体,难以通过色谱法除去。紧接着,用LiBH4将rac–16还原为醇rac–17,从而获得第二个关键的起始原料,即扩环反应。原位形成的三氟乙酸盐被苄胺以分子内方式取代,形成叠氮基中间体,再经开环即可合成六元环化合物,反应混合物用饱和NaHCO3水溶液淬灭,并通过硅胶色谱纯化,从而获得目标化合物rac–18。同时,较长的反应时间对于高收率至关重要,而在使用较高温度(微波)下,会导致产生不希望的五元环异构体以及较低的转化率。为避免手性色谱的分离,作者筛选了21种脂肪酶,用于rac–18的乙酸乙烯酯进行立体定向酶促酯化反应,反应结果表明,QLM脂肪酶可选择性地获得高对映体纯度的19和(+)-18(>95% ee)。
紧接着,在NaOH的THF/MeOH(2:1)溶液中,获得定量收率的19。同时,双脱苄基比预期的困难,通过大量条件的优化后,当使用Li和EtOH的Birch条件下,可将18中内酰胺氮上的苄基选择性地除去,然后在标准条件下进行氢化可去除另一个苄基,从而获得77%收率的21。该反应需将Bn改为Boc保护基的原因有两个:首先,在随后的Mitsunobu反应中避免与叔胺的相互作用;其次,在引入羟胺基后氢化是不合适的。在Mitsunobu条件下形成4-硝基苯甲酸酯,然后在MeOH中用K2CO3皂化,可完成21中3-位立体中心的转化,经色谱法可获得68%收率的23。用N-(苄氧基)-2-硝基苯磺酰胺(24)制成的Mitsunobu条件19形成25,在Boc脱保护和色谱分离后,以51%的收率得到26。随后,在标准条件下,用Nosyl脱保护基可以形成84%收率的27。通过将光气溶液(注射器泵)缓慢添加到27中形成环状脲,并在色谱和结晶后得到81%收率的关键中间体28(通过HPLC可将纯度提高至99.6%,并将ee提高至100%)。而在Pd/C上用H2进行脱苄基反应获得定量收率的29,再经三氧化硫吡啶配合物处理和色谱分离后,可获得85%收率的30。最后,通过阳离子交换,即可获得IID572(收率为73%,纯度>95%)。
总结
作者开发了一种新型β-内酰胺酶抑制剂IID572的合成途径,克服了总收率低(0.9%)且关键起始底物不易获得的弊端。通过此方案,可实现从市售的底物开始,并以19步获得1.7-2.9%总产率的目标化合物IID572。该方案主要通过亚胺叶立德[3+2]环加成反应制备复杂的中间体,同时通过酶拆分方法获得所需的对映异构体(高ee值)。尽管在官能团相互转化方面存在一些合成挑战,但仍可以进行8 mol规模的放大,从而获得10 g的IID572以及各类中间体,为临床前毒理学研究和各类反应条件的优化提供了便捷。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
















No comments yet.