

本文作者 芃洋雪.
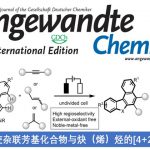
苯并咪唑酮benzimidazolone和苯并噁唑酮benzoxazolone是化学药物中的常见骨架结构,如氟哌利多droperidol、匹莫齐特pimozide、多潘立酮domperidone、氯唑沙宗chlorzoxazone等FDA批准的药物,它们的结构如图1a所示。传统的合成方法需要官能团化的苯衍生物,如邻苯基二胺的羰基化反应,2-氨基苯酚的羰基化反应,或者苯胺衍生物的环合反应等,图1b所示。但这种依赖于起始物料的合成策略限制了这类化合物的多样性合成。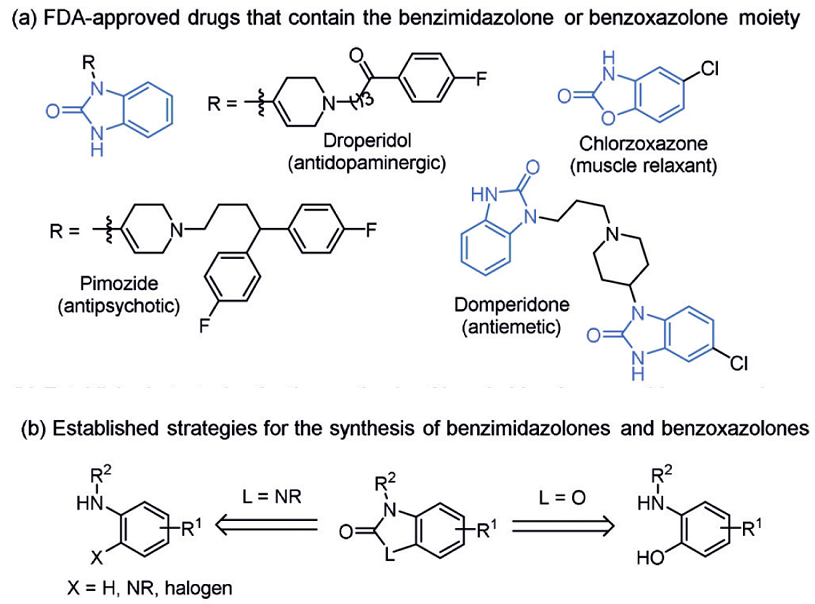
图1a含苯并咪唑酮和苯并噁唑酮的药物结构,1b合成策略.
现在,有机电化学合成逐渐引起化学家的注意,电化学合成仅需要电流就能促进氧化还原反应,不需要危险的化学试剂,而且电压可调和电极材料可以选择,能够适应于多种结构的底物,应用范围更广。近年来自厦门大学的徐海超团队,发展的电化学合成方法,解决了多种杂环的合法,如苯丙含硫杂环[1],含氮杂环[2,3],含氧杂环[4]等。
课题组主页:http://chem.xmu.edu.cn/groupweb/hcxu/
论文作者介绍:
徐海超(教授,博士生导师),1983年生于湖南耒阳
教育经历:
- 学士(厦门大学,2002-2006)
- 博士(华盛顿大学,2006–2010)
- 博士后(耶鲁大学,2011–2013)
履历:
- 副教授(厦门大学,2013–2014
- 教授(厦门大学,2014–)
- 福建省闽江学者特聘教授(厦门大学,2015–)
获奖情况:
- Chinese Chemical Society Prize for Young Scientists 2017 (2017中国化学会青年化学奖)
- The Distinguished Lectureship Award, sponsored by the Chemical Society of Japan, 2018厦门大学2018年度田昭武学科交叉奖(特等奖)
研究领域:
Organic electrochemistry 有机电化学
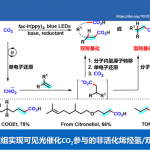
在这里,作者报道了这种全新的合成方法,一步反应同时进行苯环和五元杂环的环合得到苯并咪唑酮和苯并噁唑酮,合成策略如图1d所示。论文”De Novo Synthesis of Highly Functionalized Benzimidazolones and Benzoxazolones through an Electrochemical Dehydrogenative Cyclization Cascade”发表在Angew. Chem. Int. Ed. 2019, 58, 9017-9021, DOI:10.1002/anie.201904931,并被选为内封面VIP论文。
图1d. 全新从头设计的电化学合成.
作者首先以易得的脲类化合物1为底物,进行反应条件的筛选。反应设备是实验室最常见的三口烧瓶,加入两个电极。因化合物1对碱敏感,易发生碱催化的离子型氢胺化反应,因此反应体系未加入任何碱性试剂,而且这个底物在酸性环境下稳定。经过优化反应条件,最终确定反应温度为100℃,电流为10mA, 阳极电极材料为网状玻璃态碳RVC(Reticulated Vitreous Carbon)电极,阴极为铂板电极,1当量的三氟乙酸TFA为酸性添加剂,DMF为溶剂(entry 1),此时产物2的收率为83%。产物2的氧化电位(Ep/2=1.26V vs SCE)高于起始物料1的氧化电位(Ep/2=0.96V vs SCE),因此2不会发生氧化降解。相对于不添加TFA(entry 2)收率下降至65%,说明TFA可能通过促进阴极的质子还原从而促进反应,同时避免了碱催化的副反应。降低反应温度到50℃(entry 3),更换其他溶剂如DMA(entry 4),DMSO(entry 5),TFE(entry 6)都降低了产率。 反应温度较高有可能是底物1需要克服从trans-trans的构象转化为易于反应tans-cis构象的能障。更换阳极材料为Pt(entry 7),石墨(entry 8),阴极材料为Ni(entry 9),不锈钢(entry 10)以及RVC(entry 11),都不如RVC和Pt电极组合有效。具体反应条件和收率见表1所示。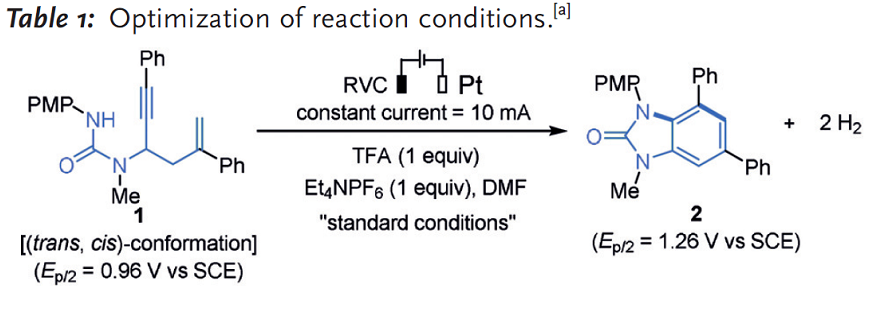

表1. 各反应条件和收率.
反应底物脲类化合物2的取代基范围很广,如图2所示。虽然大部分是烷基或芳香取代基,不论是三取代的烯烃生成苯环对位取代的苯并咪唑酮衍生物(17-23),还是全取代的产物(30-35),都有较高的收率。 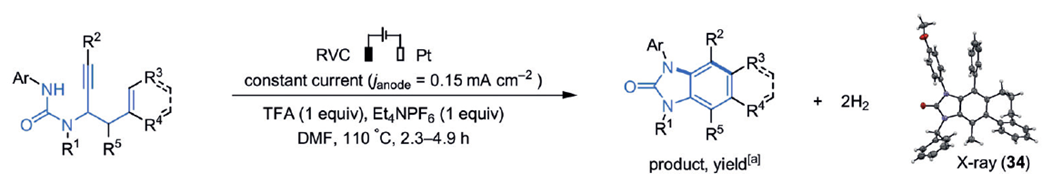
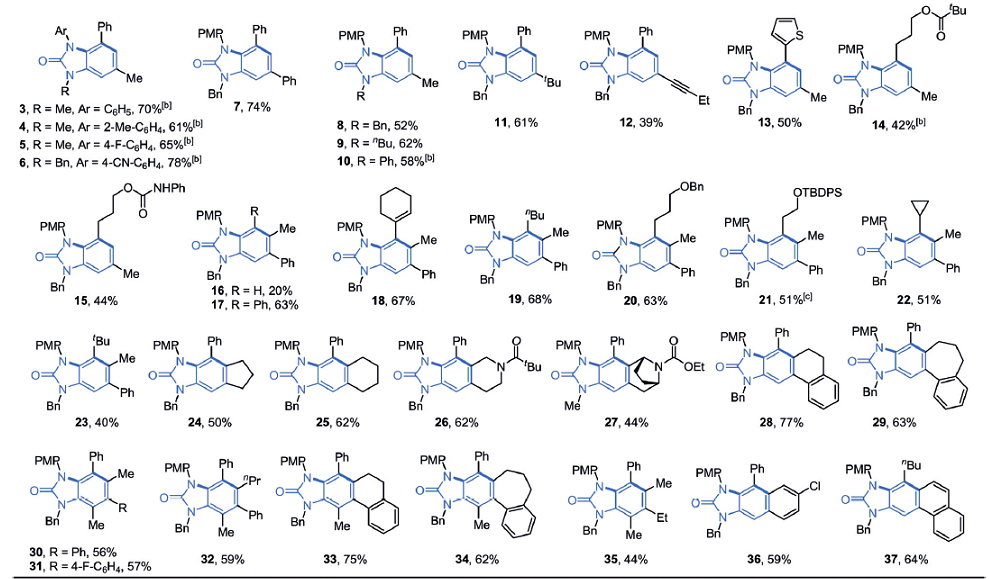
图2. 不同反应底物的产物和收率.
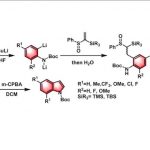
作者接下来研究了这个反应是否可以用于合成苯并噁唑酮,以丙炔基氨基甲酸酯为起始物料。沿用相同的反应条件,虽然可以得到产物,但收率很低,产物38的收率仅为18%,这可能是其中的三氟乙酸降解了丙炔基氨基甲酸酯。但在TFE(2,2,2-三氟乙醇)中,加入5当量的乙酸作为酸添加物时,加热到80℃使溶剂TFE回流时,收率提高至47%。底物同样兼容多种烷基和芳香基取代基,产物(38-47)结构和收率如图3所示。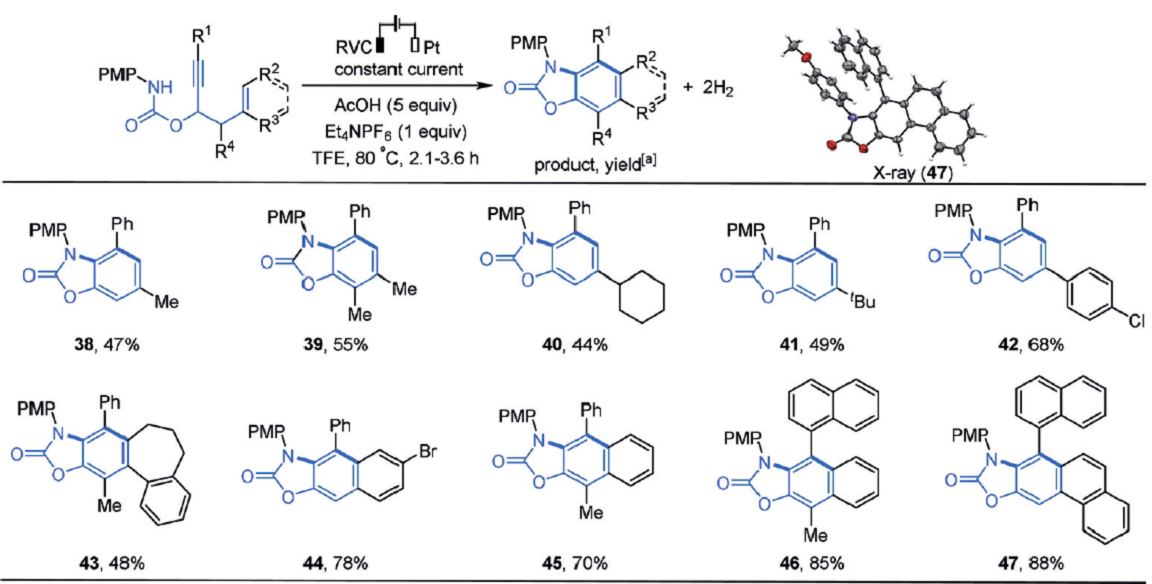
图3. 苯并噁唑酮类化合物的结构剂及合成收率.
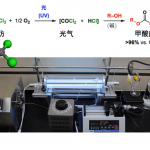
这个反应规模可以达到10g级别,反应装置如图4所示,10.4g的反应底物48可以生成5.9g的产物20,收率为57%。
图4. 电化学合成装置.
而且产物20可以继续衍生,如脱去氮上的苄基后再与POCl3反应,得到氯化产物49。20也可以和NBS反应生成溴化产物50,和I2反应生成碘化产物51。侧链上的苄氧基同样可以继续反应,脱去苄基保护基生成醇52,或者转化为乙酰氧基53和氯化产物54,结构衍生如图5所示。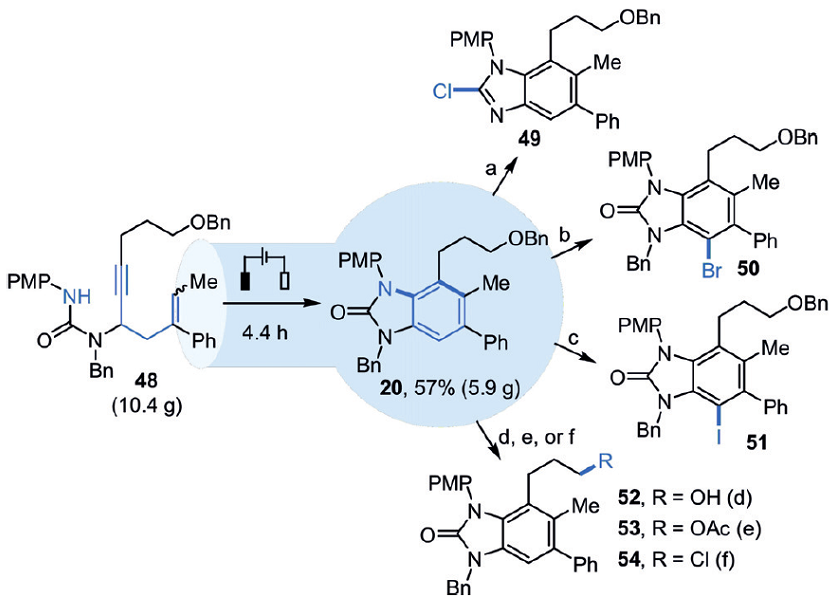
图5. 化合物20的结构衍生.
最后仍然是关键的机理研究。根据之前的研究,作者曾发现二茂铁作为中等强的的氧化还原催化剂在电催化中生成酰胺自由基。在这个反应中加入二茂铁,55转化为20%的环己二烯56和痕量的41,还有12%的单环产物57。在标准反应条件下,56又可以转化为41,并且收率可达67%,如图6a所示,这个现象说明可能存在自由基串联反应。 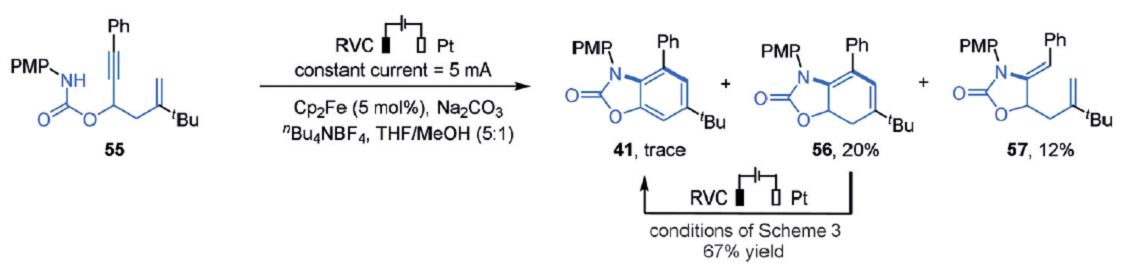
图6a. 二茂铁作用下的合成反应.
由此,作者推测了反应机理:起始物料的芳香胺基团在阳极首先氧化脱氢后生成酰胺自由基Ⅰ,接着发生5-exo-dig环合反应生乙烯基自由基Ⅱ。自由基Ⅱ可以发生两种不同的反应,path a 继续发生6-endo-trig环合反应生成环状碳自由基Ⅲ,或者path b 5-exo-trig环合后生成自由基Ⅳ,再通过一个三环自由基中间体Ⅴ再转化为Ⅲ。自由基Ⅲ继续发生单电子转移SET氧化,然后脱氢,得到中间体Ⅵ,最终失去两个电子和质子后得到苯并杂环产物。电子从阳极转移至阴极,同时和质子结合生成H2,如图6b所示。
图6b. 可能的反应机理.
总结:
作者开发的电化学合成苯并咪唑酮和苯并噁唑酮的方法,通过脱氢还原串联反应, 能够以较高的收率得到多官能团修饰的产物,并且不需要额外的化学试剂。通过简单的搭建装置,这种方法可以达到10g规模的合成,具有很好的实用性。
参考文献:
- Xu, F.; Long, H.; Song, J. Xu, H.-C. Angew. Chem. Int. Ed. 2019, 58, 6650.-6653. DOI:10.1002/anie.201901610
- Wu, Z.-J.; Li, S.-R., Xu, H.-C. Angew. Chem. Int. Ed. 2018, 57,14266-144070. DOI:10.1002/ange.201807683
- Hou, Z.-W.; Mao, Z.-Y., Melcamu, Y. Y.; Lu, X., Cu, H.-C. Angew. Chem. Int. Ed. 2018, 57, 1636-1639. DOI:10.1002/anie.201711876
- Cai, C.-Y., Xu, H.-C. Nat. Commun. 2018, 9, 3551. DOI: 10.1038/s41467-018-06020-8
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
















No comments yet.