
本文作者:石油醚
有机化学是一门既充满艺术又充满科学的基础学科,兼具艺术性和科学性,神秘而富有魅力的学科。在我们的衣食住行中,都影响这我们。那一个有机反应是如何发生的呢,中间有经历了哪些复杂的变化?带着这些疑问,化学家开始研究反应过程中的变化,并提出了有机反应机理的概念。1903年,亚瑟·拉普沃斯(Arthur J. Lapworth)通过研究安息香缩合反应1,2,提出了第一个有机反应机理。上一期,我们介绍了反应动力学分析和FTIR与NMR技术在有机反应机理中的应用。本期将带大家了解DFT计算(Figure 1)和自由基检测手段应用于有机反应机理的研究中的实例。
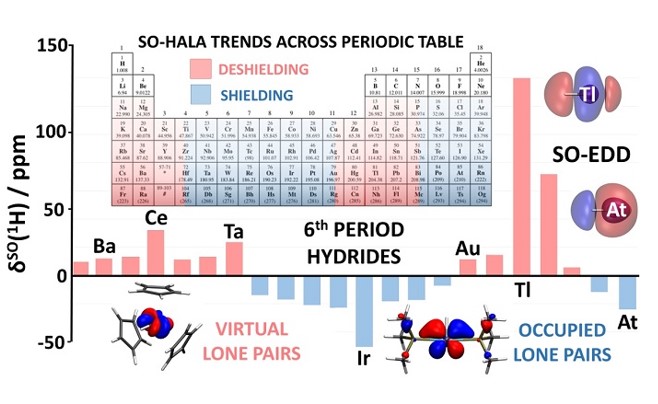
Figure 1 DFT计算
1.密度泛函数(DFT计算)
化学是一门既充满艺术又充满科学的基础学科,兼具艺术性和科学性,神秘而富有魅力的学科。在我们的衣食住行中,都影响这我们。随着时代,计算机技术,密度泛函数理论的成熟,我们翻开不同的化学相关的期刊搜寻文献的时候,都会发现DFT被用来:1)预测分子结构,指导目标分子的合成;2)利用计算化学模型实现分子催化剂从无到有的设计,精准定向合成工业反应的催化剂,3) 预测化学反应的过渡态并提出可能的反应机理,4)挖掘一切可能的化学信息,来创造下一个突破获神奇的材料,5)研究表面化学转化最激动人心的方面来揭示催化循环的真实动力学等众多方面的应用。量子化学计算方法对实验的指导性越来越高,其重要性在诺奖中也有充分体现。(Figure 2)

Figure 2 量子化学与诺贝尔化
密度泛函理论(DFT)为研究多原子体系中的电子基态能量提供了一个通用的框架。
其历史可以上溯到由Thomas和Fermi 在1920年代发展的Thomas-Fermi模型,Thomas-Fermi模型是很重要的第一步,但是由于没有考虑Hartree-Fock理论指出的原子交换能,Thomas-Fermi方程的精度受到限制。1928年保罗·狄拉克在该模型基础上增加了一个交换能泛函项。Slater 用Xɑ法进一步阐述了这些构想,最后,直到六十年代中期,Kohn及其他研究人员才共同奠定了现代理论的基础(Ohenberg-Kohn(HK)变分原理和Kohn-Sham(KS)方程)(Figure 3)。自那时起,特别是在过去的二十年中,密度泛函理论在电子结构问题中的应用有了很大幅度的增长。现在,密度泛函理论已经成了研究凝聚态与复杂分子环境中电子结构计算基本原理最常用的方法之一3-5。目前,DFT在构型计算、电子分布、电荷处理等方面有着较广阔的用途。基于DFT的软件有:应用较广的是商业软件VASP和Gaussian,以及开源免费的Quantum ESPRESSO;容易上手的是MS的集成软件CASTEP和dmol3;至于SIESTA(学院用户开源,免费)、pwscf等则是各有特色。然而中国的理论化学起步于1950年,是由唐敖庆先生开创的。从1950年开始到现在,国内涌现了一批计算化学方面的专家,学科也有了长足的发展,为以后国家理论化学的发展奠定了长足的发展。而DFT计算在我们有机化学方法学领域中也有了长足的发展,对于我们理解反应机理和动力学的发展提供了强大的理论基础。
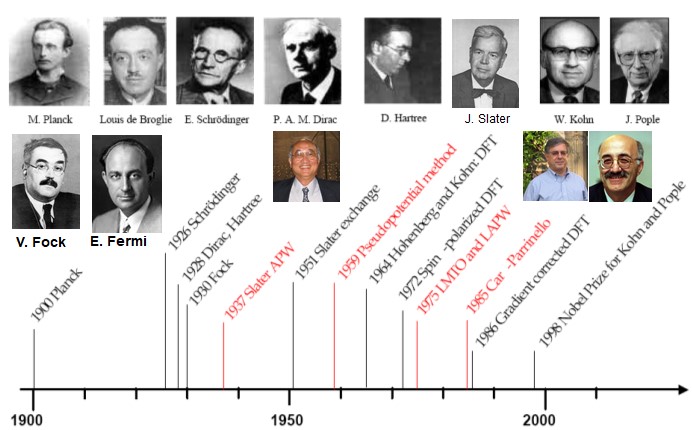
Figure 3 DFT发展历史(图来自科塔学术)
去对称化策略可以通过简单易得的前手性或内消旋原料出发,通过控制反应位点或者中心,来构建季碳手性中心。南方科技大学的刘心元教授利用金属催化的不对称自由基化学开展系统性研究工作6-9。2020年在《Nature Catalysis》发表了题为“Catalytic enantioselective desymmetrising functionalization of alkyl radicals via Cu(I)/CPA cooperative catalysis”( 铜/手性磷酸催化的烷基自由基的不对称去对称化官能团化反应)(doi: 10.1038/s41929-020-0439-8 ),使用去对称化不对称自由基1,2-氧氟烷基化反应,得到了具有多个手性中心的四氢呋喃骨架化合物。该反应的非对映选择性和对映选择性,同时反应条件温和,底物适用范围广泛。(Scheme 2)
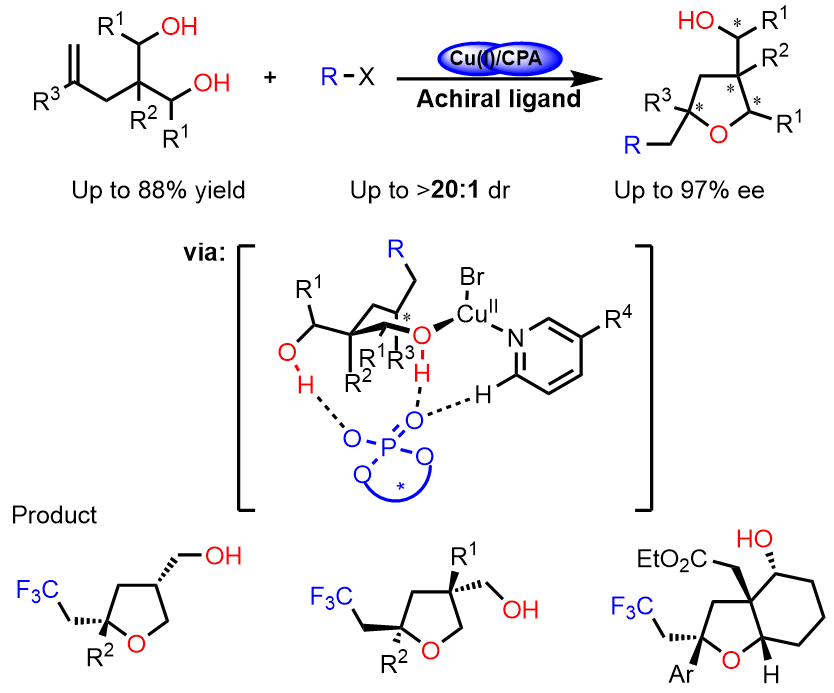
Scheme 2 铜/手性磷酸催化的烷基自由基官能化
刘心元教授授与浙江大学的洪鑫教授合作,使用DFT计算来对铜/手性磷酸催化的烷基自由基的不对称去对称化官能团化反应机理做了深入的研究。在DFT计算之前作者结合前人的工作和自己见解首先对选择性形成C-O键可能的途径进行了预测10-12。(scheme 3)途径A烷基取代的自由基直接攻击Cu键合的烷氧基,反应其他两个途径涉及SET过程的碳正离子中间体(途径B)或自由基捕获的烷基铜(III)中间体,然后进行还原消除获得产物(途径C)。

Figure 3 对映选择形成C–O键的途径
DFT计算的C-O键形成过程的自由能曲线(Figure 4)。DFT计算表明:从苄基自由基中间体Int12结合Cu(II) –CPA物种Int13,生成Cu(III)中间体Int14,该中间体是由Fig.3c中path C路径提出的Cu(III)物种。由于与二醇的多个氢键相互作用,使得Int14中CPA阴离子并非直接与Cu配位。从Int14开始,我们采取经典的方法去研究C-O还原消除的过渡态,尽管付出了巨大的努力,我们仍无法找到这样的过渡态。取而代之的是确定了一种非常简便的逐步还原消除的途径。 Cu(III)中间体Int14进过过渡态TS15进行C–Cu键的裂解,从而生成苄基阳离子的Cu(I)中间Int16。中间体Int16的发现对于提出的SET过程(Fig.3c,path B)的有很大的作用。随后通过过渡态TS17形成C–O键会产生去对称化产物,并逐步再生Cu(I)催化剂。因此,整个C-O键的形成是一个逐步过程,涉及的Cu(III)物种(Fig.3c,path C)和Cu(I)物种(Fig.3c, path B)。除了Cu(II)物种Int13之外,Figure 4中的含铜中间体和过渡态均为单线态物种。 Int16的三重态非常不利,这排除了涉及自由基取代的路径(Fig.3c,path A)
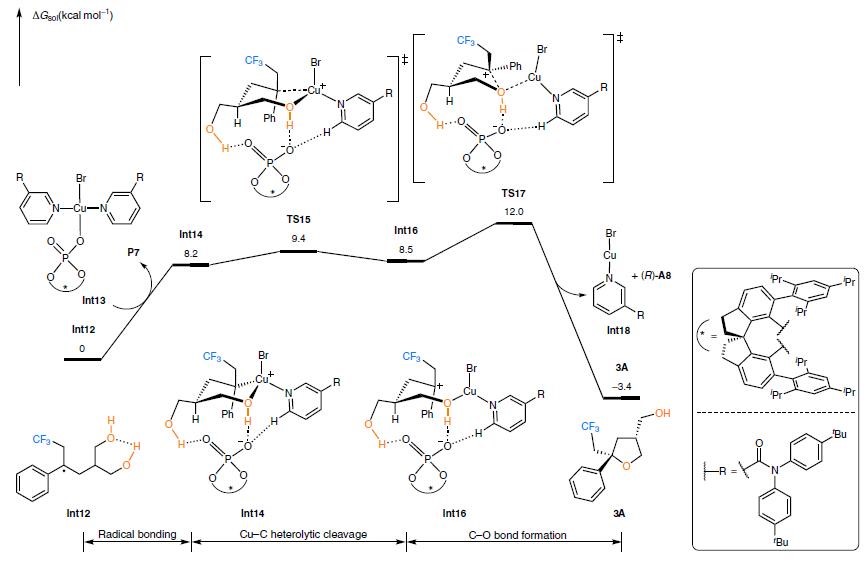
Figure 4 DFT 计算C-O键形成过程的自由能曲线
DFT计算的机理模型为理解立体选择性的因素奠定了扎实的基础13-16。C–O键形成过渡态的优化结构和能量如Fig.5所示。对C-O键形成过渡态进行了的构象搜索发现,TS17比其他三个过渡态(TS17-a,TS17-b和TS17–c)最小的少于3.5 kcal mol-1,这与实验中优异的对映选择性和非对映选择性是一致的。对比TS17和TS17–a,配位的路易斯碱和底物的苯基之间的π–π堆积的相互作用决定了苄基位置的立体选择性,基于相互作用片段的计算以及IGM分析确认这种了π-π堆积的相互作用。由于苯基位于吡啶的远端,TS17-a中就不存在上面所述的稳定π-π堆积相互作用。路易斯碱充当将CPA阴离子的手性转移至远端苄基立体中心的桥梁作用。第二个立体选择性控制因素是二醇的氢键和CPA阴离子之间的作用。在最优的过渡态TS17中,两个羟甲基是相对的,由于生成的是三级立体中心,TS17-b和TS17-c的构象不利于这两个过渡状态的形成。

Figure 5 C-O键形成过渡态的优化结构和能量
刘心元教授授与浙江大学的洪鑫教授合作,使用DFT计算对铜/手性磷酸催化的烷基自由基的不对称去对称化官能团化反应机理做了深入的研究发现手性磷酸负离子与底物中的两个羟基形成的氢键网络以及非手性吡啶配体与底物上的芳环之间的π–π相互作用在产物的立体选择性控制中起到了至关重要的作用。
参考文献
- [1] Lapworth, A. XCVI.—Reactions involving the addition of hydrogen cyanide to carbon compounds. J. Chem. Soc., Trans. (1903) 83, 995-1005, doi:10.1039/CT9038300995.
- [2] Lapworth, A. CXXII.—Reactions involving the addition of hydrogen cyanide to carbon compounds. Part II. Cyanohydrins regarded as complex acids. J. Chem. Soc., Trans. (1904) 85, 1206-1214, doi:10.1039/CT9048501206.
- [3] Froese Fischer, C. General Hartree-Fock program. Comput. Phys. Commun. (1987) 43, 355-365, doi:https://doi.org/10.1016/0010-4655(87)90053-1.
- [4] Born, M. & Oppenheimer, R. Zur Quantentheorie der Molekeln. Ann. Phys. (1927) 389, 457-484, doi:10.1002/andp.19273892002.
- [5] Kohn, W. & Sham, L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. (1965) 140, A1133-A1138, doi:10.1103/PhysRev.140.A1133.
- [6] Phipps, R. J., Hamilton, G. L. & Toste, F. D. The progression of chiral anions from concepts to applications in asymmetric catalysis. Nat. Chem. (2012) 4, 603-614, doi:10.1038/nchem.1405.
- [7] Sibi, M. P. & Porter, N. A. Enantioselective Free Radical Reactions. Acc. Chem. Res. (1999) 32, 163-171, doi:10.1021/ar9600547.
- [8] Choi, J. & Fu, G. C. Transition metal–catalyzed alkyl-alkyl bond formation: Another dimension in cross-coupling chemistry. Science (2017) 356, eaaf7230, doi:10.1126/science.aaf7230.
- [9] Cherney, A. H., Kadunce, N. T. & Reisman, S. E. Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents To Construct C–C Bonds. Chem. Rev. (2015) 115, 9587-9652, doi:10.1021/acs.chemrev.5b00162.
- [10] Lin, J.-S. et al. A Dual-Catalytic Strategy To Direct Asymmetric Radical Aminotrifluoromethylation of Alkenes. J. Am. Chem. Soc. (2016) 138, 9357-9360, doi:10.1021/jacs.6b04077.
- [11] Zhu, R. & Buchwald, S. L. Enantioselective Functionalization of Radical Intermediates in Redox Catalysis: Copper-Catalyzed Asymmetric Oxytrifluoromethylation of Alkenes. Angew. Chem. Int. Ed. (2013) 52, 12655-12658, doi:10.1002/anie.201307790.
- [12] Gephart, R. T. et al. Reaction of CuI with Dialkyl Peroxides: CuII-Alkoxides, Alkoxy Radicals, and Catalytic C–H Etherification. J. Am. Chem. Soc. (2012) 134, 17350-17353, doi:10.1021/ja3053688.
- [13] Reid, J. P., Simón, L. & Goodman, J. M. A Practical Guide for Predicting the Stereochemistry of Bifunctional Phosphoric Acid Catalyzed Reactions of Imines. Acc. Chem. Res. (2016) 49, 1029-1041, doi:10.1021/acs.accounts.6b00052.
- [14] Duarte, F. & Paton, R. S. Molecular Recognition in Asymmetric Counteranion Catalysis: Understanding Chiral Phosphate-Mediated Desymmetrization. J. Am. Chem. Soc. (2017) 139, 8886-8896, doi:10.1021/jacs.7b02468.
- [15] Wheeler, S. E. Understanding Substituent Effects in Noncovalent Interactions Involving Aromatic Rings. Acc. Chem. Res. (2013) 46, 1029-1038, doi:10.1021/ar300109n.
- [16] Lefebvre, C. et al. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys (2017) 19, 17928-17936, doi:10.1039/C7CP02110K.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!




















No comments yet.