
前面介绍了K. C. Nicolaou教授的结构鉴定过程和第二次全合成工作,这次我们来接着介绍D. A. Evans教授在2007年的ent-Azaspiracid-1全合成工作[1]。
-
A. Evans 全合成研究
1). 逆合成分析
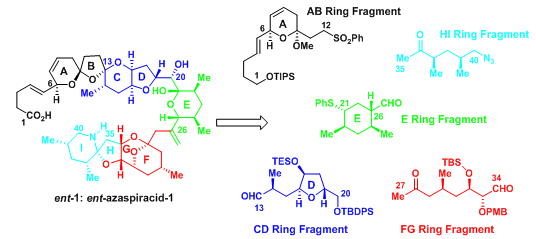
Evans教授根据之前合成altohyrtin C[2]和bryostatin 2[3]的合成经验,在C-20位设计了醛96与α-烷氧基磺酸盐97的偶联反应,但在C-20位的立体选择性却有很大的困难,于是考虑羟基可以通过先氧化后还原的方法得到希望的构型,在最后的合成中有所体现。醛96的螺环同理可以从之前的文献找寻得用Lewis酸催化形成[4];99通过官能团转化反应可得到98,而99可通过碎片100和101反应得到[5]。97可由通过热力学控制下的一系列成环反应得到;102则通过aldol反应得到。

图1:Evans’Retrosynthetic Analysis of ent-Azaspiracid-1
2). ent-Azaspiracid-1的全合成
A:ABCD环体系的合成
如图2所示,烯丙醇115的合成是由107和110两个片段拼接而成的。在107的合成中,丙烯酮105先与苯硫酚加成得到的硫醚106接着与丙二烯格式试剂反应得到高炔丙醇107;底物108与109在Grubbs-Hoveyda Ⅱ 的催化下发生烯烃复分解反应得weinreb酰胺110;两当量丁基锂处理的高炔丙醇107与110反应得到的112经羟基保护后得113,由于weinreb酰胺的配位稳定作用,羰基不会被过度还原;113经114所示过渡态,经历CBS Reduction得到115[6]。

图2: Evans‘ Synthesis of Allylic Alcohol 115
烯丙醇115在Lindlar催化剂作用下被选择性还原成顺式烯烃,接着用TBSCl保护羟基得116;弱碱性条件下脱除乙酰基后氧化此羟基得酮117;117先脱TBS保护基后在酸性条件下关A环得苯硫醚118;118被氧化得到砜119;119本是当初设想的偶联碎片,但在反应时产率不高,于是将酯基还原成羟基并用Si保护,看似无关紧要的变化,改变了距离砜基的距离,这在后面的反应中可以看出,对产率有较大影响。
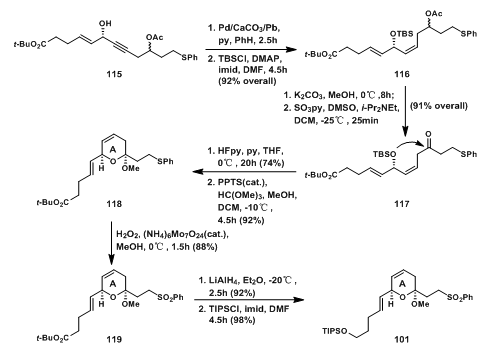
图3: Evans‘ Synthesis of A-Ring Sulfone 101
在D环碎片100的合成中首先运用到了不对称的 hetero-ene reaction代替aldol reaction得到高烯丙醇124,123与120的两个氧配位形成的不稳定平面导致了反应的高选择性;酯基被weinreb酰胺取代,羟基用PMB保护得weinreb酰胺125;发生lithium-halogen exchange的126与125反应脱去胺部分后得127;127中的羰基经Felkin-Anh Reduction[7]高选择性的得到醇129;在第一步得到的烯烃经臭氧氧化得到酮,随后在酸的催化下羟基与形成的羰基反应关上D环,形成缩酮131,随后脱除甲氧基后得到期望的反式四氢呋喃异构体,其中此反应的立体选择性对立体环境有细致的要求[8],当羟基为PMB基团保护时,对立体控制有重要作用;随后脱除PMB基团,羟基用TES重新基团保护得到缩酮133,再经过单电子还原剂LiDBB脱除苄基保护[9],最后经Parikh-Doering氧化得到D环碎片100。
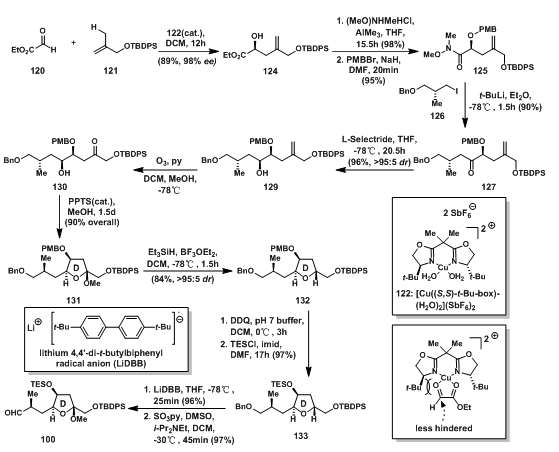
图4: Evans‘ Synthesis of D-Ring Aldehyde 100
101在LDA的作用下脱质子后与醛100反应得四种异构体99[5],随后将形成的羟基经Dess-Martin Oxidation氧化成羰基,并在还原条件下脱除砜基得单一化合物98;TBAF脱除Si保护后关C环,再经PPTS(cat.)催化形成B环,得到螺缩酮136;最后先脱除Si保护后将羟基氧化成醛,得ABCD环体系96。
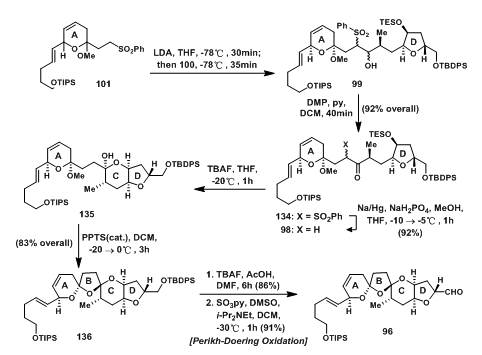
图5: Evans‘ Synthesis of ABCD-Ring Aldehyde 96
B:EFGHI环体系的合成
在EFGHI环体系的合成中,Evans教授课题组的箱式催化剂(box catalyst)也应用其中。137和138在箱式锡催化剂(tin Ⅱ box catalyst)催化下发生Mukaiyama Aldol Reaction,高选择性的生成反式aldol产物141[10, 11],Cu的d电子层电子为d9,可以形成平面的配位形式,而Sn的d层电子全充满,故采取四面体式的配位方式,如140所示。为了减小呋喃氧与醛氧的电性排斥作用,采用140所示的方式,高选择性得到141;还原α,β-不饱和内酯的碳碳双键,羟基用PMB保护得内酯143;143经还原开环[12]得到的羟基随后用TBS保护,最后脱除一个羟基上的保护,得一级醇145;一级醇145先与TsCl反应的OTs基,随后被氰基取代得146;Boc酸酐与二级胺反应的产物随后被硼氢化锂还原成为一级醇148;再将醇用TMS保护后,甲基锂还原氰基,并在后处理中一并脱除TMS保护基得到的酮149;最后149的羟基经Parikh-Doering氧化,被氧化成醛基得103。
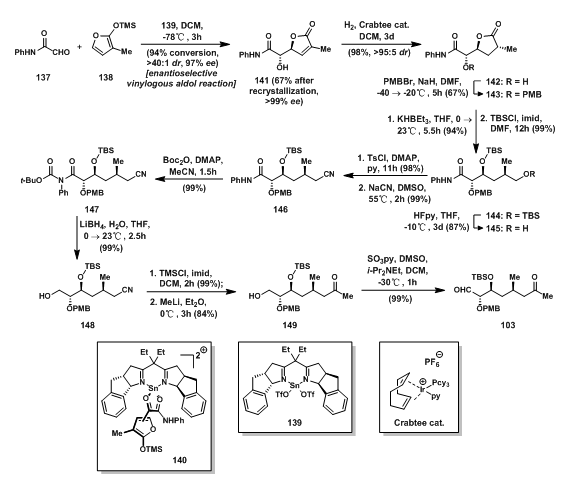
图6: Evans‘ Synthesis of Aldehyde Fragment 103
150和151在二价铜箱式催化剂(Cu(Ⅱ) box catalyst 152)催化下发生不对称的氧杂D-A反应(hetero-Diels-Alder[32])高选择性得到二氢吡喃产物154,由153可以看出,为消除平面上的位阻影响,150选择从下面反应,得到产物;二氢吡喃154从位阻更小的面被还原得到四氢吡喃155;四氢吡喃在酸催化下与苯硫酚反应,得到构型转变的O,S-杂缩酮156;随后在碱催化下发生平衡反应,得到酯基反转的更有利产物157,最后酯基被还原成醛基得E环碎片醛104。
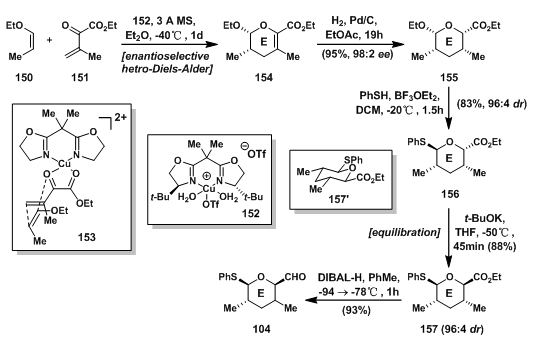
图7: Evans‘ Synthesis of E-Ring Aldehyde Fragment 104
在合成碎片ent-33的合成中,是以中间产物55为起始原料的,55先用Si氢脱除乙氧基得到的158再用四氢铝锂还原得到一级醇159,然后将羟基用I置换,得到碘化物160;碘化物先进行lithium-halogen交换得过渡态161,随即开环得到烷氧负离子,随后被TsCl捕获得甲苯磺酸盐162;162发生Wacker Oxidation,双键被氧化成羰基,得到酮163;163的OTs基被叠氮取代得到ent-33;ent-33,103及104需要用两个aldol反应将其链接起来,接上而来,ent-33先生成烯基硅醚164,随后与103发生Mukaiyama Aldol反应,经过中间过渡态165,得到aldol产物166。
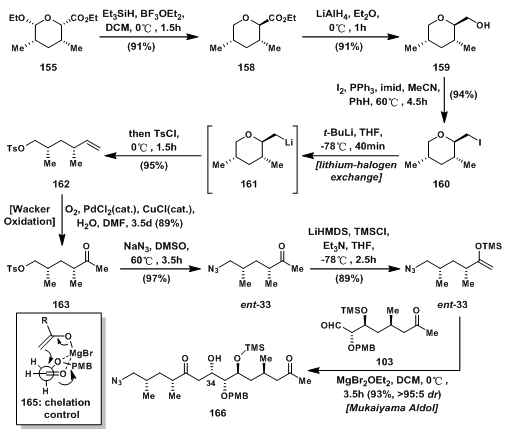
图8: Evans’ Synthesis of Methyl Ketone ent-33, and Its Coupling with Aldehyde 103
另外一个aldol反应是甲基酮166与醛104发生的Paterson Aldol反应。甲基酮166先去质子后与硼结合生成烯醇式,随后与醛104发生反应得到102,值得注意的是,这里的选择性并不是很好(60:40 dr),这是缺少可以协助反应的官能团参与反应导致的。实际上,这种直接的策略对这4个C=O双键的反应性进行了仔细考量,在精心设计的路线下,才有可能完成3个碎片的拼接。
碎片偶联产物102在HF中脱Si保护基后,同时发生GF环的环化,得到EFG环化合物167;用Dess-Martin氧化,将仲羟基氧化成羰基后,DDQ脱除PMB保护基得169;在Pd/C,H2中,叠氮被还原成瞬时的氨基,立刻发生环化反应,得HI环170;中间体170的氨基先被Teoc保护基保护后,随后在Tebbe Reagent[14]的催化下,羰基转换成双键,得到烯烃171,最后将苯硫醚氧化成砜97。
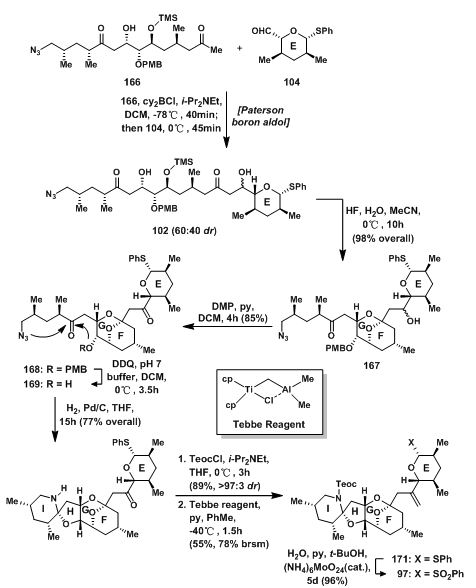
图9: Evans‘ Synthesis of EFGHI-Ring Sulfone 97
C:碎片连接及全合成
如图10所示,α-烷氧基砜97先被丁基锂去质子,随后与醛96发生sulfone-aldehyde反应[5]得到中间过渡态172,用NaOAc/AcOH处理,使砜基脱去以及氧负离子得质子,得到27% 173和23% 174;C-20位的构型可通过氧化还原的方法扭转,于是173先发生Swern Oxidation,后用四氢铝锂还原得到期望的174;TBAF脱保护基得到的175经两步氧化(Dess-Martin Oxidation & Pinnick Oxidation),将羟基转变成羧基,得到产物ent-Azaspiracid-1,完成全合成。
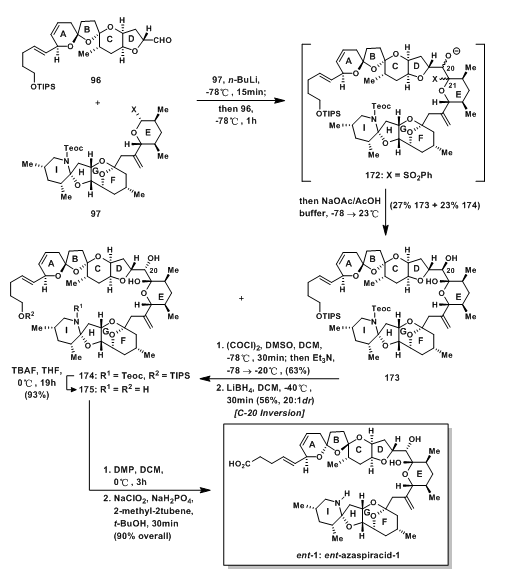
图10:Final Stages and Sompletion of Evans‘ Total Synthesis of ent-Azaspiracid-1
-
总结
复杂分子的全合成在对azaspiracid的探索中起着重要的作用,这不仅是在对其结构的测定中,对生理活性研究也提供了客观的供应量。在这其中的一些合成工作也为新的天然产物结构鉴定提供了新方法,也为探索这些生物毒素的作用机理提供了帮助。正如前面章节所讲,全合成在化学和生物鉴定新的天然产物中,特别实在缺少足够的天然产物原料时,起着至关重要的作用,。
参考文献
- a) D. A. Evans, L. Kværnø, J. A. Mulder, B. Raymer, T. B. Dunn, A. Beauchemin, E. J. Olhava, M. Juhl, K. Kagechika, Angew. Chem. Int. Ed., 2007, 46, 4693; b) D. A. Evans, L. Kværnø, J. A. Mulder, B. Raymer, T. B. Dunn, A. Beauchemin, E. J. Olhava, M. Juhl, K. Kagechika, Angew. Chem. Int. Ed., 2007, 46, 4698; c) D. A. Evans, L. Kværnø, T. B. Dunn, A. Beauchemin, B. Raymer, J. A. Mulder, E. J. Olhava, M. Juhl, K. Kagechika, D. A. Favor, J. Am. Chem. Soc., 2008, 130, 16295;
- (a) A. Evans, B. W. Trotter, B. Côté, P. J. Coleman, Angew. Chem. Int. Ed. Engl. 1997. 36, 2737. DOI: 10.1002/anie.199727381 (b) D. A. Evans, B. W. Trotter, B. Côté, P. J. Coleman, Angew. Chem. Int. Ed. Engl. 1997. 36, 2741. DOI: 10.1002/anie.199727411 (c) D. A. Evans, B. W. Trotter, B. Côté, P. J. Coleman, Angew. Chem. Int. Ed. Engl. 1997. 36, 2744. DOI: 10.1002/anie.199727441 (d) David A. Evans, B.Wesley Trotter, Paul J. Coleman, Bernard Côté, Luiz Carlos Dias, Hemaka A. Rajapakse, Andrew N. Tyler, Tetrahedron, 1999, 55, 8671. DOI.org/10.1016/S0040-4020(99)00438-X;
- (a) A. Evans, P. H. Carter, E. M. Carreira, J. A. Prunet, A. B. Charette, M. Lautens,Angew. Chem. Int. Ed. 1998, 37, 2354. DOI: 10.1002/(SICI)1521-3773(19980918)37 (b) D. A. Evans, P. H. Carter, E. M. Carreira, J. A. Prunet, A. B. Charette, M. Lautens, J. Am. Chem. Soc., 1999, 121. 7540. DOI: 10.1021/ja990860j;
- Margaret A. Brimblea, Farès A. Farèsb, Tetrahedron, 1999, 55, 7661 org/10.1016/S0040-4020(99)00387-7;
- (a) L. Field, J. Am. Chem. Soc., 1952, 74. 3919. DOI: 10.1021/ja01135a062. (b) L. Field, J. W. McFarland, J. Am. Chem. Soc., 1953, 75. 5582. DOI: 10.1021/ja01118a031;
- J. Corey, C. J. Helal, Angew. Chem. Int. Ed. 1998, 37, 1986. DOI: 10.1002/(SICI)1521-3773(19980817)37;
- For a review on models for stereoinduction, see: A. Mengel, O. Reiser, Chem. Rev., 1999, 99, 1191. DOI: 10.1021/cr980379w;
- H. Larsen, B. H. Ridgway, J. T. Shaw, K. A. Woerpel, J. Am. Chem. Soc., 1999, 121, 12208. DOI: 10.1021/ja993349z;
- (a) P. K. Freeman, L. L. Hutchinson, J. Org. Chem., 1980, 45, 1924. DOI: 10.102 1/jo01298a034. (a) P. K. Freeman, L. L. Hutchinson, J. Org. Chem., 1983, 48, 4705, DOI: 10.1021 /jo00172a049;
- For a review on Mukaiyama aldol reaction, see: Mukaiyama, T. Angew. Chem. Int. Ed. 2004, 43, 5590. doi:10.1002/anie.200300641;
- A. Evans, M. C. Kozlowski, J. A. Murry, C. S. Burgey, K. R. Campos, B. T. Connell, R. J. Staples, J. Am. Chem. Soc., 1999, 121, 669. DOI: 10.1021/ja9829822;
- For a review on metal hydride reductants, see: H. C. Brown, S. Krishnamurthy, Tetrahedron, 1979, 35, 567. org/10.1016/0040-4020(79)87003-9;
- For a review on asymmetric hetero-Diels-Alder cycloaddition of carbonyl componds, see: H. Pellissier, Tetrahedron, 2009, 65, 2839. org/10.1016/j.tet.2009.01.068;
- Tebbe, F. N.; Parshall, G. W.; Reddy, G. S. J. Am. Chem. Soc., 1978, 100, 3611. DOI:10.1021/ja00479a061;
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!





















No comments yet.