

本文作者:alberto-caeiro
松原 诚二郎(Matsubara Seijiro),日本有机化学家,日本京都大学大学院工学研究科教授。图片:实验室介绍。
经历
- 1977–1981 京都大学 工学部 工業化学科(指導教官:野崎 一 教授)
- 1981–1983 京都大学大学院 工学研究科 材料化学専攻 修士課程(指導教官:野崎 一 教授)
- 1983–1986 京都大学大学院 工学研究科 材料化学専攻 博士課程(指導教官:野崎 一 教授)
- 1984洛桑联邦理工学院(EPFL)博士課程(指導教官:Manfred Schlosser 教授)
- 1986–1995 京都大学 工学部 工業化学科 助手(内本 喜一朗 教授)
- 1988–1989 斯坦福大学(Stanford University)博士研究員(Barry M. Trost 教授)
- 1995–2006 京都大学大学院 工学研究科 助教授(大嶌 幸一郎 教授)
- 2006–現在 京都大学大学院 工学研究科 教授
获奖经历
- 1987 第3回 井上研究奖励賞
- 1998 有機合成化学協会 有機合成化学奖励賞
- 2014 Asian Core Program Lectureship Award 2014 (Malaysia)
- 2014 Asian Core Program Lectureship Award 2014 (Korea)
- 2017 日本化学会 第34回学术賞
工作介绍
1. 有机催化[1]
Matsubara教授开发了有机催化剂催化的杂-Michael加成反应以实现不对称构建各类杂环化合物,和不对称反应构造轴手性中心。通过设计不同活化模式,他们实现了有机催化的高对映选择性转化。
1)分子内不对称杂-Michael加成反应
分子内杂-Michael加成反应是可以用于直接构建杂环化合物的。但由于该反应速度快,反应难以实现高的对映选择性。为了解决这个问题,Matsubara教授使用具有酸碱双功能催化剂(如氨基硫脲,磷酸等),通过氢键实现多点协同增效,实现了高对映选择性的分子内杂-Michael加成反应。在反应中,如果不同时激活多个位点,反应就无法进行,而该多点激活的策略为手性控制提供了有效的手性环境。
如下图所示,通过该策略,分子内杂-Michael加成反应[2,3]都可以高对映选择性地实现。

Highly enantioselective intramolecular hetero-Michael addition reaction
2)轴手性化合物合成
Matsubara教授设想可通过将双官能有机催化剂识别的分子构象转化为轴向手性的构建,从而扩展有机小分子催化剂的用途。该想法在异喹啉N-氧化物的溴化反应中被验证是可行的[4]。如下图所示,通过苯酚的溴化反应,以奎宁衍生物为有机催化剂,高对映选择性地实现了轴手性的构建。
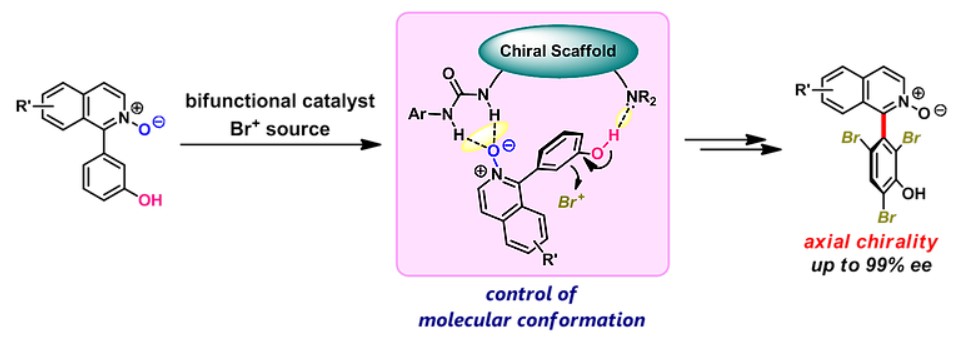
Enantioselective Synthesis of Axially Chiral Isoquinoline N-Oxides
3)α,β-不饱和羧酸衍生物的Michael加成反应
α,β-不饱和羧酸衍生物由于含有多个可与有机催化剂作用的杂原子位点,其不对称杂-Michael加成反应的对映选择性较差。Matsubara教授采用涉及共价相互作用的多点激活策略,利用手性亲核催化剂与羧酸衍生物的加成-消除反应生成的α,β-不饱和酰基铵物种作为关键中间体,实现了高对映选择性的sulfa-Michael 加成环化反应[5]。如下图所示,通过该方法,可高对映选择性的合成药物中常见结构1,5-苯并噻氮平。
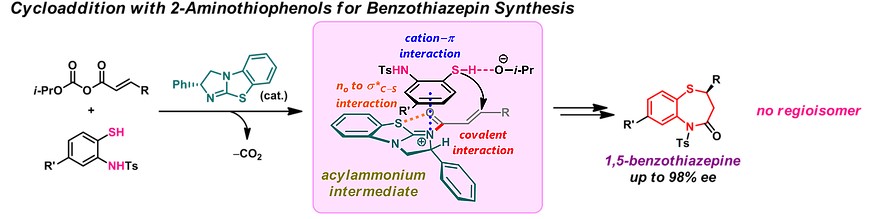
Enantioselective sulfa-Michael reaction of α,β-unsaturated carboxylic acid derivatives
2. 过渡金属催化的反应
1)经C-X键断裂的C-C成键反应
如下图所示,Matsubara教授利用过渡金属的反应特性,将廉价易得的杂环化合物中的C-X键(X = O, N)切断,再与活性物种炔烃反应,得到了更为复杂的杂环化合物,实现了杂环化合物的高附加值转化[6]。
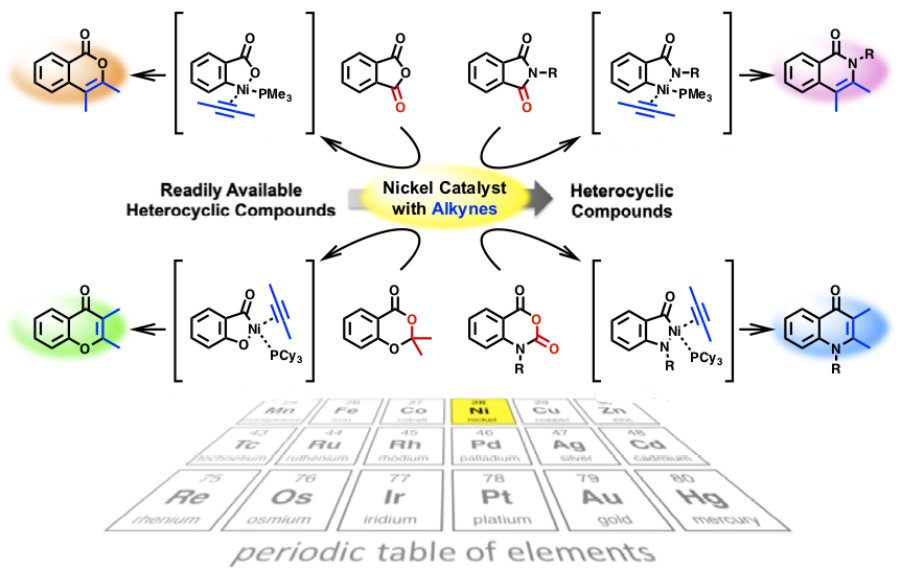
Nickel catalyzed high value-added chemical transformations
此外,Matsubara教授还实现了碘苯与炔烃反应得到插烯的烯基碘化合物[7]。

Nickel-catalyzed intermolecular carboiodination of alkynes with aryl iodides
2)经零价镍氧化环合的C-C成键反应[8]
Matsubara教授利用零价镍容易氧化环合的特性,通过调控配体,实现了α,β-不饱和羰基化合物和炔烃的各类氢转移和环化反应{9}。逆电子流向的D-A反应也可以通过该过程实现,在该反应中,可得到氧化环合中间体单晶结构[10]。
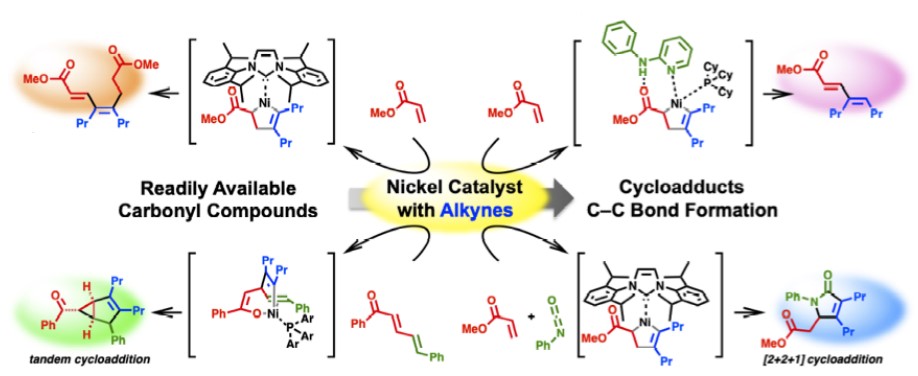
Ni-Catalyzed C–C Bond Formation via oxidative cyclization
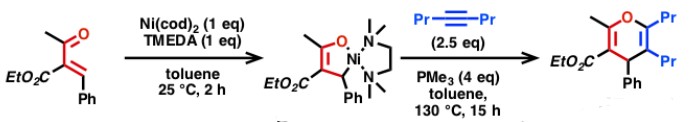
Nickel-Catalyzed [4 + 2] Cycloaddition of Enones with Alkynes
Matsubara教授通过零价镍和卡宾配体IPr的催化体系,实现了伯醇和炔烃氧化还原中性的氢转移反应[11],反应是通过伯醇脱氢形成的醛与炔烃经过氧化环合中间体进行。
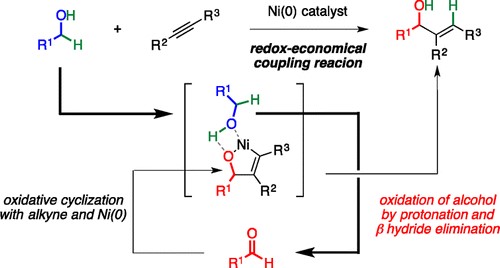
Nickel catalyzed redox-neutral coupling of alcohols and alkynes
参考文献
- [1] a. Asano, K.; Matsubara, S. ACS Catal. 2018, 8, 6273. doi.org/10.1021/acscatal.8b00908; b. Asano, K. J. Synth. Org. Chem., Jpn. 2016, 74, 1194–1205.
- [2] Oxo-Michael addition reaction: a. Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2011, 133, 16711. doi.org/10.1021/ja207322d; b. Yoneda, N.; Fujii, Y.; Matsumoto, A.; Asano, K.; Matsubara, S. Nat. Commun. 2017, 8, 1397. doi.org/10.1038/s41467-017-01099-x; c. Yoneda, N.; Fukata, Y.; Asano, K.; Matsubara, S. Angew. Chem., Int. Ed. 2015, 54, 15497. doi.org/10.1002/anie.201508405.
- [3] Aza-Michael addition reaction: a. Fukata, Y.; Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2013, 135, 12160. doi.org/10.1021/ja407027e; b. Miyaji, R.; Asano, K.; Matsubara, S. Org. Lett. 2013, 15, 3658. doi.org/10.1021/ol401538b.
- [4] a. Miyaji, R.; Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2015, 137, 6766. doi.org/10.1021/jacs.5b04151; b. Wada, Y.; Matsumoto, A.; Asano, K.; Matsubara, S. RSC Adv. 2019, 9, 31654. doi.org/10.1039/C9RA05532K.
- [5] a. Fukata, Y.; Asano, K.; Matsubara, S. J. Am. Chem. Soc. 2015, 137, 5320. doi.org/10.1021/jacs.5b02537; b. Fukata, Y.; Yao, K.; Miyaji, R.; Asano, K.; Matsubara, S. J. Org. Chem. 2017, 82, 12655. doi.org/10.1021/acs.joc.7b02451.
- [6] a. Matsubara S. J. Am. Chem. Soc. 2008, 130, 6058. doi.org/10.1021/ja7114426; b. Matsubara S. J. Am. Chem. Soc. 2008, 130, 17226. doi.org/10.1021/ja806569h; c. Matsubara S. J. Am. Chem. Soc. 2009, 131, 7494. doi.org/10.1021/ja900805y; d. Matsubara S. J. Am. Chem. Soc. 2011, 133, 11066. doi.org/10.1021/ja203829j; e. Matsubara S. J. Am. Chem. Soc. 2013, 135, 13636. doi.org/10.1021/ja4068172.
- [7] Matsubara S. Chem. Commun. 2018, 54, 12580. doi.org/10.1039/C8CC07560C.
- [8] Matsubara S. Bull. Chem. Soc. Jpn. 2014, 87, 1058. doi.org/10.1246/bcsj.20140158.
- [9] a. Matsubara S. Chem. Commun. 2010, 46, 7229. doi.org/10.1039/C0CC01754J; b. Matsubara S. Chem. Commun. 2010, 46, 8055. doi.org/10.1039/C0CC02613A; c. Matsubara S. Chem. Commun. 2011, 47, 2658. doi.org/10.1039/C0CC04061D; d. Matsubara, S. Angew. Chem., Int. Ed. 2011, 50, 8956. doi.org/10.1002/anie.201104286.
- [10] a. Matsubara S. J. Am. Chem. Soc. 2009, 131, 1350. doi.org/10.1021/ja807952r; b. Matsubara S. Chem. Commun. 2011, 47, 6150. doi.org/10.1039/C1CC10890E.
- [11] Matsubara S. J. Am. Chem. Soc. 2014, 136, 7797. doi.org/10.1021/ja500666h.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!

























No comments yet.