
本文作者:杉杉
导读
近日,南方科技大学贾铁争教授课题组和宾夕法尼亚大学Marisa C. Kozlowski教授课题组共同在自然通讯(Nature Communications)杂志上发表论文,报告了一种无过渡金属催化的现芳基甲基亚砜和醇的交叉偶联反应,从而获得多种芳基醚衍生物。同时,也对两种药物分子中关键中间体进行了制备。
Transition-metal-free formal cross-coupling of aryl methyl sulfoxides and alcohols via nucleophili cactivation of C-S bond
Guolin Li, Yexenia Nieves-Quinones, Hui Zhang, Qingjin Liang, Shuaisong Su, Qingchao Liu,Marisa C. Kozlowski &Tiezheng Jia
Nat. Commun. ASAP DOI: 10.1038/s41467-020-16713-8
正文
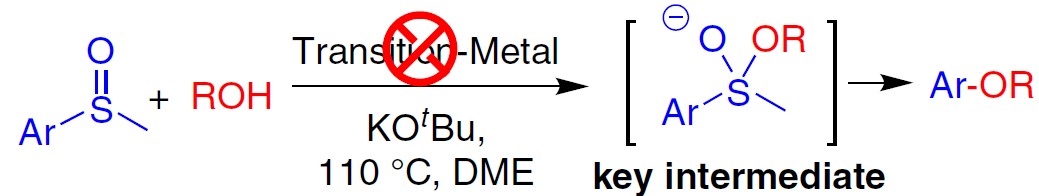
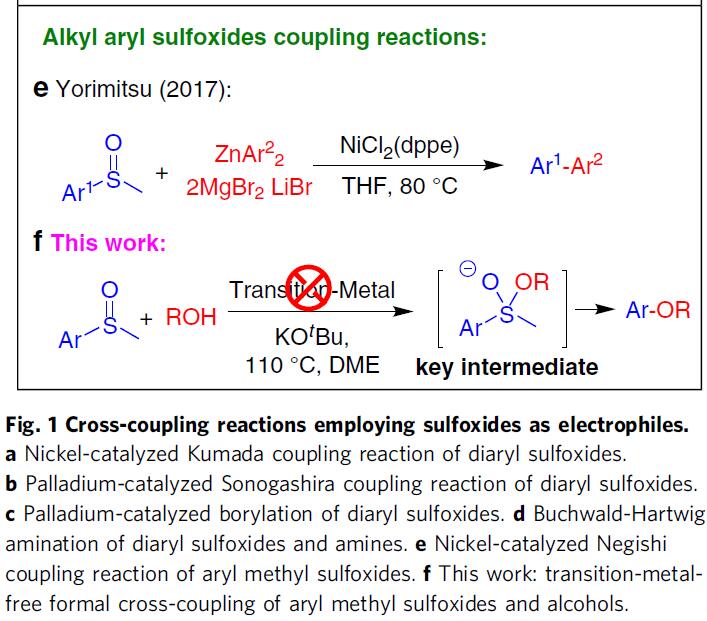
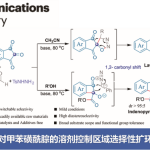
在过去的几十年中,交叉偶联反应已成为研究的热点,但对于亲电底物的进一步扩展(常规为芳基卤化物、磺酸盐或碳酸盐),仍具有一定的挑战。由于有机硫化合物化学稳定性、结构通用性等特点,在交叉偶联中使用有机硫化合物值得研究。迄今为止已报道了多种利用芳基硫醚或砜的交叉偶联反应,如众所周知的Liebeskind-Srogl交叉偶联反应。然而,在过渡金属催化经C-S键活化的交叉偶联反应中,亚砜作为亲电子试剂的应用仍然很少(Fig. 1)。
1979年,Wunkert课题组首次使用二芳基亚砜作为亲电子试剂,在镍催化下,实现Kumada偶联反应(Fig. 1a)。2017年,Yorimitsu课题组使用二芳基亚砜作为亲电试剂,在钯催化下,实现Sonogashira偶联反应(Fig. 1b)。同时,该课题组报道了一种钯催化剂,该催化剂可促进二芳基亚砜与二硼烷化合物的双硼化(Fig. 1c)。然而,该反应存在收率低、官能团耐受性有限等的问题。近期,该课题组又报道了一种N-杂环卡宾钯配合物的催化剂,实现了二芳基亚砜和胺之间的Buchwald-Hartwig胺化反应(Fig. 1d)。然而,芳基甲基亚砜的收率非常低。此外,Yorimitsu课题组也在2017年首次以烷基芳基亚砜作为亲电试剂,经镍催化实现C-S键的氧化加成,这与Negishi偶联程序兼容,从而得到联芳基产物(Fig. 1e)。然而,存在自偶联副产物与所需的偶联产物的分离难题。
通常,在过渡金属催化下,亚砜与亲核试剂的交叉偶联反应涉及C-S键的氧化加成。同时,过渡金属催化剂可能需要复杂的配体,这可能会大大增加花费。此外,在制药和其他应用中去除痕量过渡金属仍然是一个挑战。相反,无过渡金属的交叉偶联策略可以克服上述问题。此外,受Martin课题组在无金属催化剂下,可将硫代或亚砜基选择性转化为烷氧基的启发,在此,南方科技大学贾铁争教授课题组和宾夕法尼亚大学Marisa C. Kozlowski教授课题组共同报告了一种无过渡金属催化的交叉偶联策略,可实现芳基甲基亚砜与醇的反应,从而获得多种烷基芳基醚衍生物(Fig. 1f)。

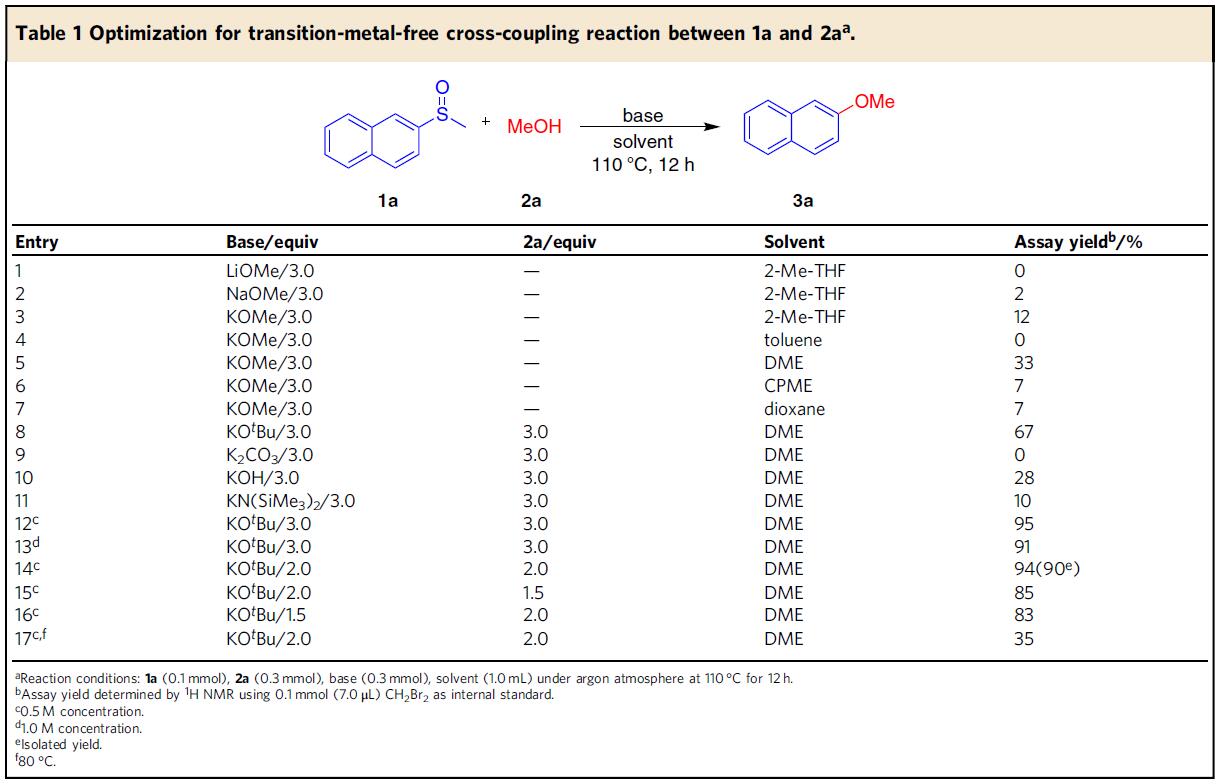
首先,作者以2-萘基甲基亚砜1a作为模型底物,进行了反应条件的筛选(Table 1)。筛选结果表明,当以KOtBu作为碱,使用1a为亲电试剂(1eq),甲醇2a为亲核试剂(2eq),可在110℃下于DME(0.5 M)中反应12h,可获得最佳效果。
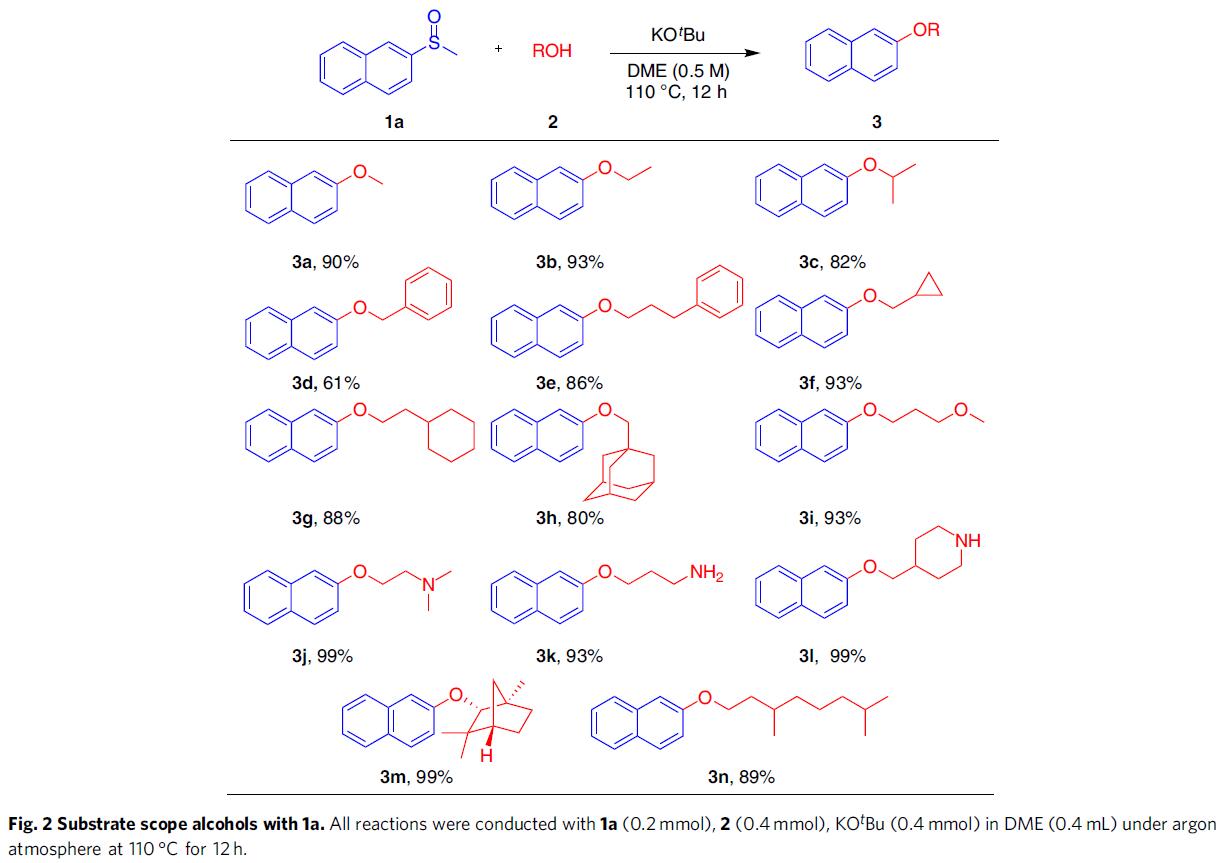
在获得上述最佳反应条件后,作者开始对其他脂肪醇2的底物范围进行了扩展(Fig. 2)。甲醇、乙醇、异丙醇,均以良好的收率获得相应的产物3a–3c。在α-或γ-位具有苯基取代的脂肪醇,也具有良好的耐受性(3d和3e)。同时,具有环状的脂肪醇,甚至空间位阻更大的1-金刚烷甲醇,均可以获得较高收率的产物3f–3h。此外,具有醚或胺官能团的脂肪醇也具有良好的耐受性,并以优异的收率获得所需产物3i–31。然而,伯胺或仲胺底物不适合该体系。通过对天然产物的相关修饰,从而以优异的收率获得3m和3n。
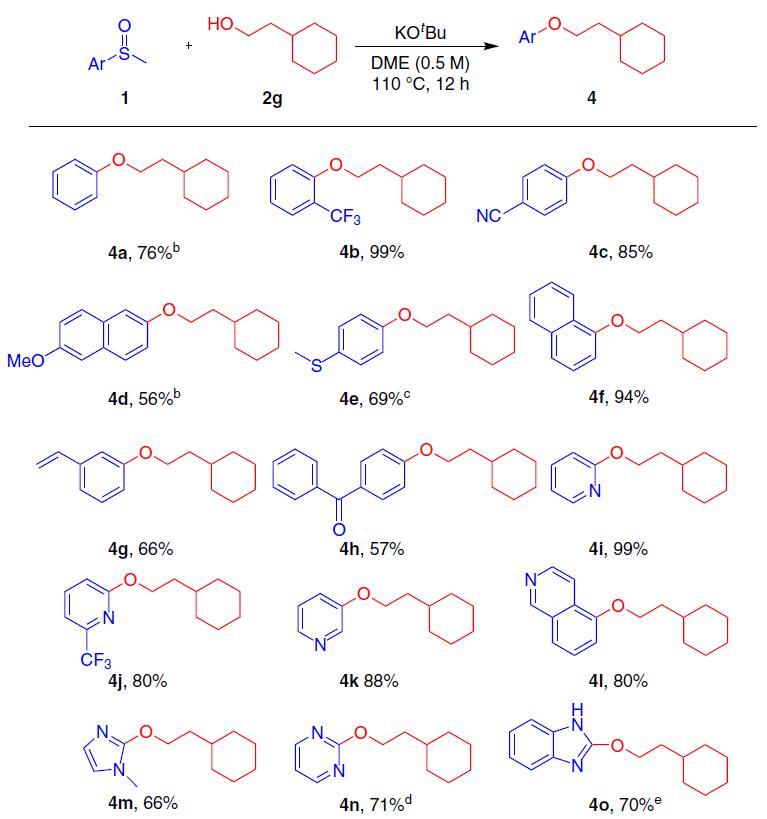
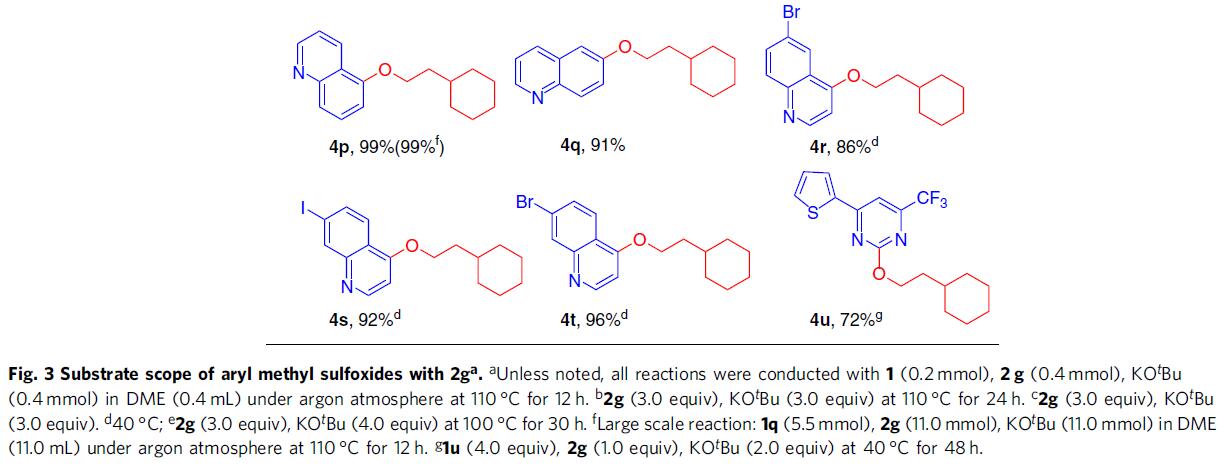
随后,作者亚砜底物范围进行了扩展(Fig. 3)。带有吸电子基(如氰基或三氟甲基)或供电子基的底物时,可获得相应的产物4b–4e,但给电子基团反应缓慢,收率偏低。值得注意的是,与先前过渡金属催化剂报道的相反,该反应对于硫甲基几乎没有反应性,从而允许获得4e,体现了独特的化学选择性。含有萘基、乙烯基或与羰基的化合物,也与体系兼容,获得相应的产物4f–4h。同时,杂芳基也具有良好的相容性,从而获得66-99%收率的产物4i-4t,并且含有溴或碘取代基不受影响,为后期偶联反应提供了多种可能。此外,该反应可直接对具有抗凋亡生物活性的化合物1v进行衍生化,从而以72%的收率获得产物4u 。
紧接着,作者进一步证明该反应在药物化学和药物开发中的应用价值(Fig. 4)。鞘氨醇1-磷酸受体调节剂6e(目前正处于治疗多发性硬化的II期临床试验中),可通过5步合成,总收率为20%。值得注意的是,在整个途径中都没有使用过渡金属,从而简化了药物的纯化过程。此外,使用高度化学选择性的过渡金属偶联策略作为关键步骤,仅需3个步,即可产生另一种获得专利的化合物,阿片类药物δ受体激动剂7c,总收率为61%。

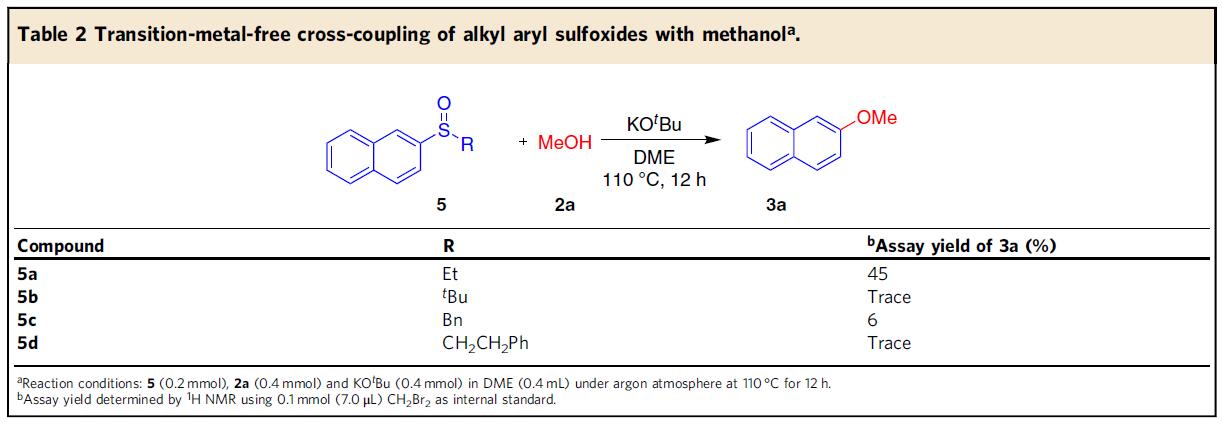
从原子经济的角度来看,芳基甲基亚砜是最有吸引力的亚砜偶联底物。尽管如此,还探索了其他烷基芳基亚砜(Table 2)。尽管3a的收率较低,但5a也作为合适的偶联底物。但5b–5d仅生成痕量的3a,可能是由于形成了亚硫酸根阴离子导致。
为了进一步了解反应的机理,作者进行了相关的对照实验(Table 3)。在标准条件下,加入TEMPO时,3a的收率没有受到影响,从而说明反应不涉及自由基途径。当使用LiOtBu或NaOtBu代替KOtBu时,3a的收率大幅降低,从而说明钾阳离子的关键作用(entries 1-2)。此外,将2.0当量的18-crown-6加入反应中,3a的收率急剧下降至5%(entry 3)。而向LiOtBu或NaOtBu的反应体系中加入2.0当量的氟化钾,反应恢复了活性,分别获得63%和27%的收率产物3a(entries 4-5)。上述结果表明,钾阳离子对该反应至关重要。

根据上述的实验和相关文献的查阅,作者提出了几种可能的反应机理(Fig. 5)。第一种机理涉及芳基甲基亚砜和醇盐之间的SNAr反应,该反应由钾离子和芳环之间的π-阳离子相互作用引发,然而钾离子更易与亚砜的氧原子配位,因而排除该可能。(Fig. 5a)。第二种机理涉及醇的协同加成消除过程,形成五元中间体使其电荷离域,但与实验结果不符,因而排除了该可能(Fig. 5b)。第三种机理涉及钾离子和亚砜氧及芳环配位,活化烷氧负离子对亚砜硫原子进行的亲核加成。随后,该氧原子与亚砜领近的芳环碳环化,再脱除DME和次磺酸钾从而获得醚化产物。该结果与计算结果一致,钾离子参与反应的活化能要低于钠离子和锂离子(Fig. 5c)。

总结
南方科技大学贾铁争教授课题组和宾夕法尼亚大学Marisa C. Kozlowski教授课题组共同报道了一种独特的无过渡金属参与的交叉偶联策略,利用芳基甲基亚砜作为醇的亲电子底物,获得烷基芳基醚衍生物。同时,该反应具有良好的官能团具有良好的耐受性,并可对天然产物和药物进行相关的后期修饰。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!


















No comments yet.