
作者:石油醚
导 读
近日,美国康奈尔大学的Todd Hyster教授课题组采用非天然光酶催化策略,实现了对消旋二级硝基烷烃的不对称C-烷基化反应,获得了过渡金属催化难以得到的手性三级硝基产物,进一步拓展了酶催化反应类型的边界,对现有化学方法起到很好的互补。基于酶本身所特有的高选择性和易进化性,结合化学原理和蛋白改造,是未来实现更多非天然、普适生物催化方法的有效策略。
“Asymmetric C-Alkylation of Nitroalkanes via Enzymatic Photoredox Catalysis
Haigen Fu, Tianzhang Qiao, Jose M. Carceller, Samantha N. MacMillan, and Todd K. Hyster*
J. Am. Chem. Soc.2023, ASAP. doi: 10.1021/jacs.2c12197”
正 文
手性α-三级胺是许多药物和天然产物分子的重要结构片段,现已发展了诸多构建手性α-三级胺的催化方法。此外,手性α-三级胺也可由相应三级硝基化合物直接还原得到,但三级硝基化合物的催化不对称合成方法尚且有限,通常由过渡金属催化的烯丙基化或有机小分子催化的加成反应得到,亟待发展更多的催化方法。对二级硝基化合物进行C-烷基化反应是一类潜在的直接构建三级硝基化合物的方法,但是这类反应通常伴随着较多的硝基O-烷基化反应副反应。为解决这个难题,前人采用单电子转移机理以及特殊的亲电试剂来提高C-烷基化的化学选择性。例如,Watson课题组以烷基卤代物为亲电试剂,发展了一系列过渡金属催化的硝基化合物C-烷基化反应,随后实现了该类反应的不对称催化。然而,通过对硝基化合物不对称C-烷基化来得到手性三级硝基化合物的催化方法还未见报道。
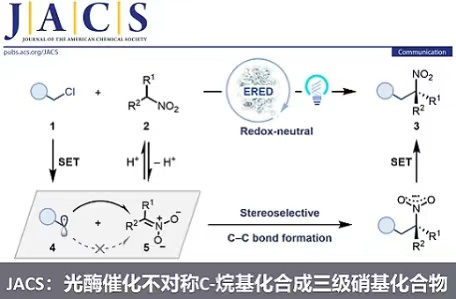
生物催化以高效、高选择性、易进化著称,美国康奈尔大学的Todd Hyster教授课题组的研究者尝试采用生物催化的方法来实现对硝基化合物的不对称C-烷基化反应。尽管已有许多酶催化的共轭加成或亨利反应可以利用nitronate(去质子化的硝基烷烃)作为亲核试剂,但没有一例可以制备手性三级硝基化合物。由于天然酶并不能催化硝基化合物C-烷基化反应,他们发展了一种全新的非天然光酶催化策略,以α-卤代羰基化合物1作为烷基化试剂,在可见光激发的烯酮还原酶(‘ene’-reductases, EREDs)催化下,高化学选择性和对映选择性地实现了对硝基烷烃的C–烷基化反应,得到了高价值的手性三级硝基化合物3(图1B)。
值得注意的是,该团队近期采用类似的非天然光酶催化策略,以α-卤代羰基化合物和α-芳香取代硝基烷烃为亲电试剂,实现了高交叉和对映选择性的Csp3–Csp3亲电偶联反应(XEC),见x-mol报道《Nature:光酶催化不对称亲电偶联》(Nature 2022, 610, 302–307)。该光酶催化XEC的关键步骤包括光诱导的选择性自由基引发、自由基加成、硝基自由基中间体发生C–N键均裂(硝基离去)、选择性氢原子转移淬灭。作者设想,如果可以通过酶来控制自由基加成的立体选择性,同时减慢硝基自由基6的C–N键均裂,加速该自由基中间体的氧化,便可得到手性三级硝基化合物3(图1B)。
首先,作者以α-氯代酰胺1a和α-苄基硝基乙烷2a为模板底物,在青蓝光(Cyan light, λmax = 497 nm)激发下,筛选了一系列烯酮还原酶,发现大多酶都可以催化反应得到消旋的三级硝基化合物3a。其中,只有来自Geobacillus kaustophilus菌株的烯酮还原酶(GkOYE)能够以56%的产率和78:22的er值得到(R)-型目标产物3a。有意思的是,当使用先前可高效催化1a和α-苯基硝基乙烷进行XEC的烯酮还原酶CsER时,也可以得到目标产物3a(85%, 50:50 er)。作者认为该化学选择性(C-烷基化vs XEC)来源于烷基取代的硝基自由基6的C–N键均裂速度较慢,从而倾向于发生单电子氧化,净反应为氧化还原中性。对照实验证实,烯酮还原酶和Cyan光对于反应性至关重要(图1C)。随后,作者基于GkOYE蛋白结构,采用迭代饱和突变策略对GkOYE进行了三轮定向进化,最终找到一个三突变体D73C/A104H/Y264W(GkOYE-G7,0.5 mol%)可高效、高立体选择性地催化反应得到3a(96%, 96:4 er, 图1D),且该反应可以放大到1.0 mmol规模(78%, 96:4 er, 196 mg)。
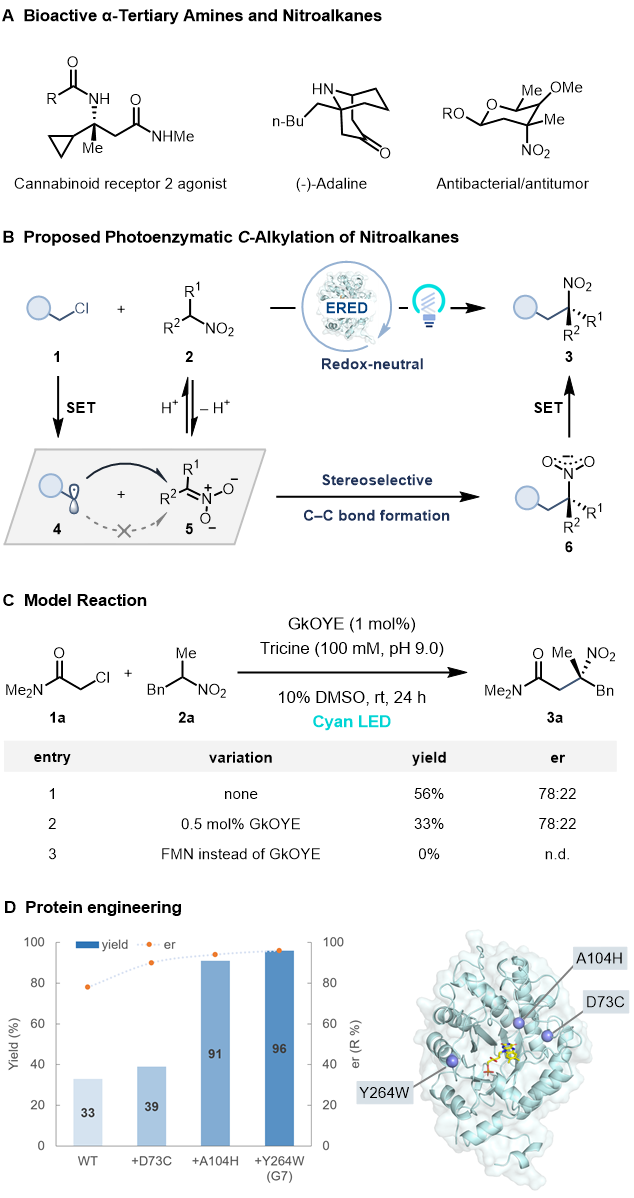
图 1.光酶催化的硝基化合物不对称C-烷基化反应。图片来源:JACS
随后,作者对该光酶催化体系的底物范围进行了探索(图 2)。多种类型的α-苄基取代硝基烷烃2a-z都可以很好地与α-氯代酰胺1a发生C-烷基化反应。如苯环的邻、间、对位可兼容带有不同电性的取代基,以56–98%的产率以及优异的对映选择性(>93:7 er)得到β-手性四取代硝基酰胺产物(7-16)。但该体系对硝基烷烃α-位取代基的体积比较敏感。此外,该体系还可以兼容不同杂环,包括富电子的噻吩和缺电子的吡啶、吡嗪取代的硝基烷烃,以91–98%的产率和高对映选择性(最高95:5 er)得到相应的杂环取代手性三级硝基化合物(20-26)。同时,直链或环状脂肪硝基烷烃也可以被该体系接受,得到相应四取代硝基产物(27-37, 22–96%),突显出该光酶催化体系广泛的底物谱。另外,多种不同类型的α-卤代羰基化合物均可被该体系兼容,包括α-氯代酰胺(如Weinreb酰胺、环状酰胺),α-卤代酮和α-卤代酯/砜(需使用CsER)均可与硝基烷烃反应得到相应三级硝基产物(32-37,图 2)。
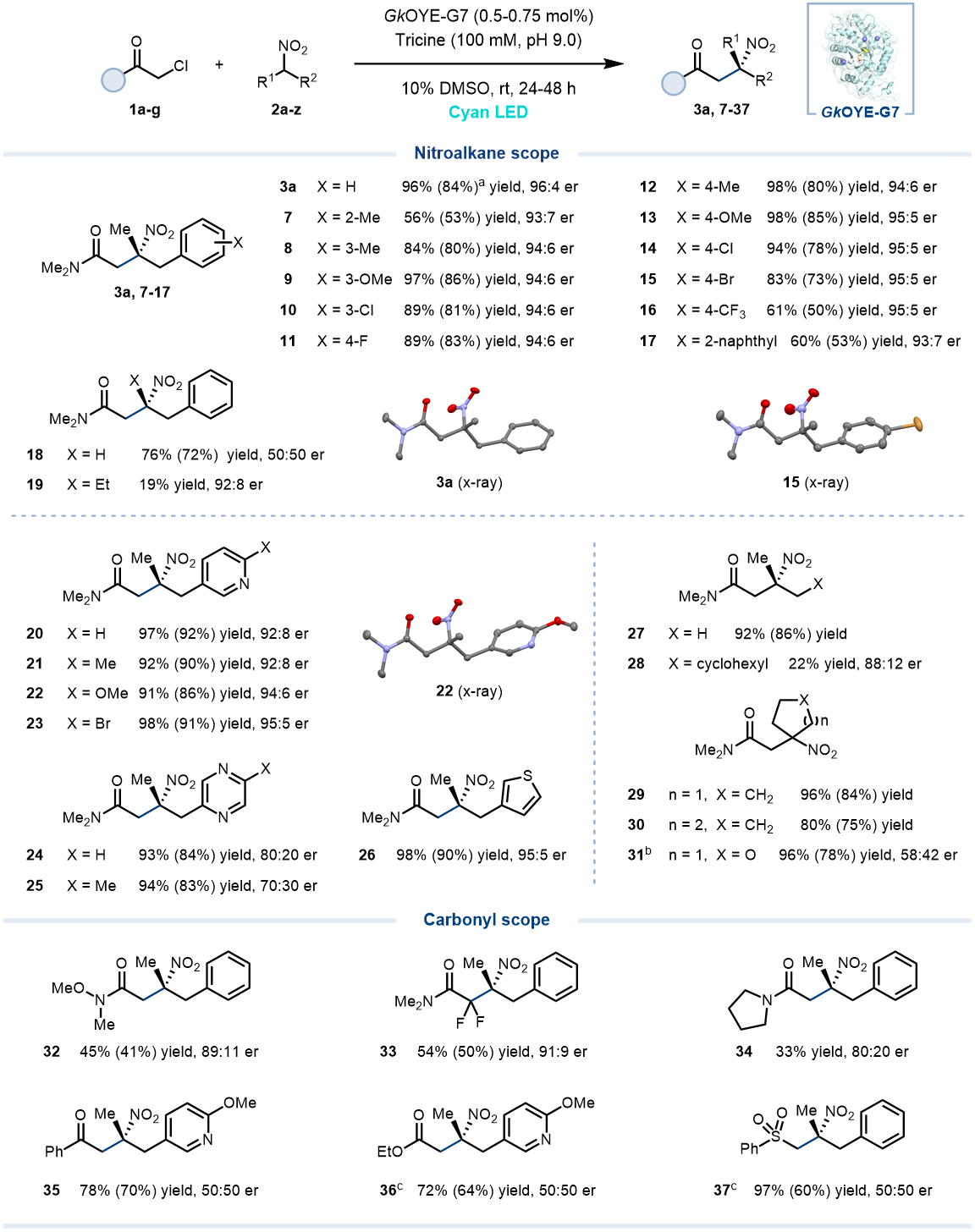
图 2. 底物范围。图片来源:JACS
对于高度进化的酶,它们通常具有很高的反应专一性。作者发现,当采用CsER来催化1a与α-芳香取代硝基烷烃38的反应时,主要得到XEC产物39b-43b(82–93%, 最高99:1 er),以及仅微量的C-烷基化产物39a-43a(b/a > 27:1,图3)。引人注目的是,当采用进化的GkOYE-G7在完全一致的条件下来催化该反应时,却得到了相反化学选择性的产物,即主要得到C-烷基化产物39a-43a(65–92%, 最高99:1 er),而几乎观察不到XEC产物39b-43b(a/b > 25:1, 图3)。到此,作者发展了两个高度专一的酶催化剂,GkOYE-G7可高效催化硝基化合物的C-烷基化,而CsER则可更好的催化XEC反应,即硝基的“留”或“去”取决于酶,充分体现了生物催化优越的化学选择性。
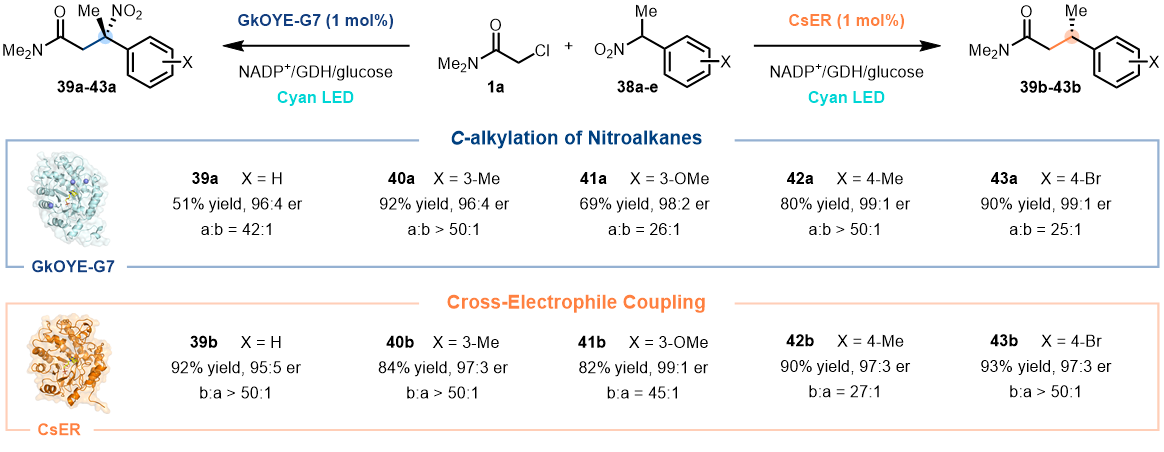
图 3. 酶控制反应性。图片来源:JACS
随后,作者设计了一条巧妙“单酶双机理”的串联反应,即原料硝基烯烃44先经由烯酮还原酶的天然氢负转移机理被还原成硝基烷烃,随后硝基烷烃再通过非天然自由基机理发生C-烷基化反应,“一酶两步”生成三级硝基化合物。以GkOYE-G7为催化剂,该串联反应具有很广的底物谱,原料硝基烯烃44全部转化为相应硝基烷烃,且以较高的产率和立体选择性得到三级硝基终产物(86–96%, 最高96:4 er, 图4A)。最后,作者对三级硝基酶产物3a进行还原,得到相应手性α-三级胺45(65%,96:4 er, 图4B)。
机理方面,作者通过UV-Vis光谱实验证实FMNhq、α-氯代酰胺1a、硝酮(nitronate模拟物)在酶的活性口袋内形成了四元电荷转移复合物(CT complex)。激发该四元复合物可实现FMNhq对1a选择性的单电子还原,得到α-酰胺自由基4,该自由基随后对nitronate 5进行立体选择性加成得到硝基阴离子自由基6,最后被FMNsq单电子氧化得到产物3(图1B)。
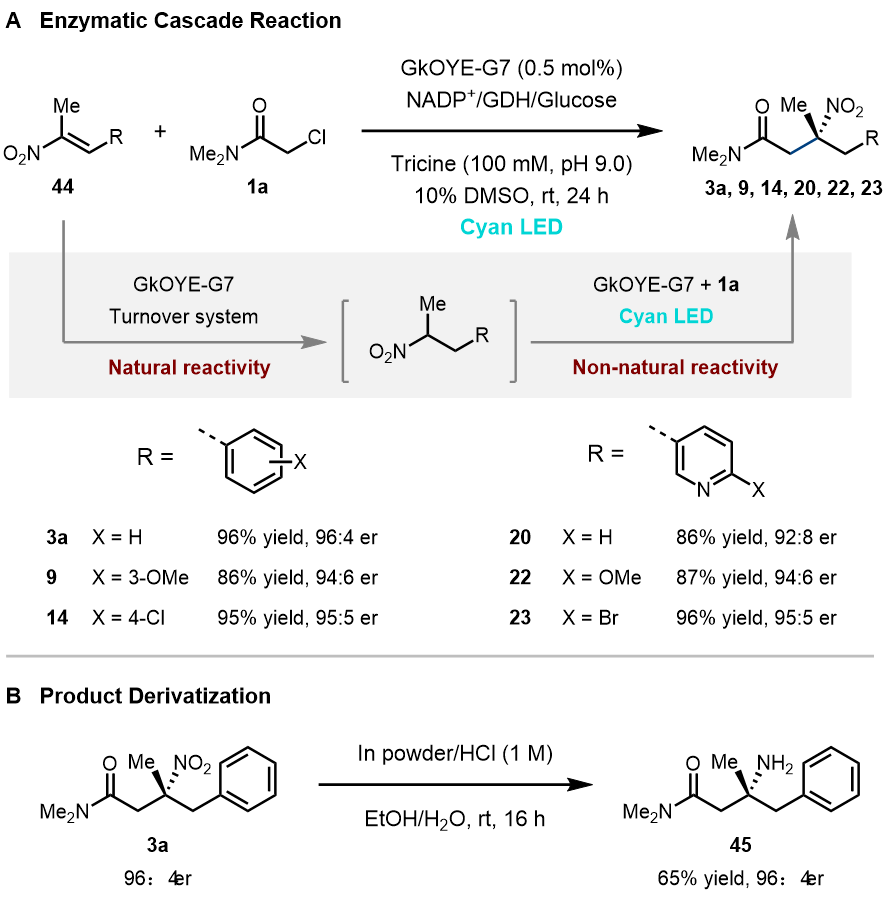
图 4. 串联反应及产物衍生化。图片来源:JACS
总之,本文采用非天然光酶催化策略,实现了对消旋二级硝基烷烃的不对称C-烷基化反应,获得了过渡金属催化难以得到的手性三级硝基产物,进一步拓展了酶催化反应类型的边界,对现有化学方法起到很好的互补。基于酶本身所特有的高选择性和易进化性,结合化学原理和蛋白改造,是未来实现更多非天然、普适生物催化方法的有效策略。相关成果在JACS上发表,康奈尔大学博士后付海根和二年级博士生乔天章为本文的共同一作,Todd Hyster教授为通讯作者。
(付海根教授供稿)

















No comments yet.