
本文作者 alberto-caeiro
瑞士苏黎世联邦理工(ETH Zürich)的Erick M. Carreira最近在JACS上报道一种消旋三级醇的不对称脱氧还原反应,反应经过配位诱导的阳离子手性控制过程。
Coordination-Induced Stereocontrol over Carbocations: Asymmetric Reductive Deoxygenation of Racemic Tertiary Alcohols”
Mayuko Isomura, David A. Petrone, Erick M. Carreira,
J. Am. Chem. Soc., 2019, 141, 4738. DOI: 10.1021/jacs.9b00862
背景
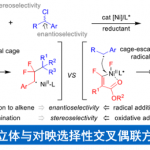
碳正离子自被欧拉教授提出并被广泛接受后,其在化学合成上被广泛应用,然而它们在不对称合成上却举步维艰。碳正离子的sp2杂化导致的两个前手性面对其不对称反应造成了极大的障碍[1](图1A左)。为此,两种方案被提出解决此问题。一种是底物手性控制(图1A中),在碳正离子旁引入手性中心,另一种是形成手性离子对(图1A右),引入手性抗衡离子控制选择性。这些方法在二级碳正离子的手性控制反应中被广泛应用,而三级碳正离子的反应仍鲜有报道。作者在前面报道过联烯醇酯的不对称烷基取代反应[2](图1B右),理论研究指出反应中存在一种亲电的η2联烯配体和一个高度离域的正电荷。这与SN1反应很相似,因此作者假设这一特性是否可以用来实现三级碳正离子的不对称反应。在这里手性中心是在与底物配位的金属上,离子化得到的碳正离子因此有了稳定的手性环境(图1C)。在1982年Siehl和Mayr的工作[3]中表明,这种联烯丙基碳正离子会在Cα和Cγ间离域(图1D)。因此在联烯与金属配位后,是否发生碳正离子的离域对反应选择性有很大的影响。
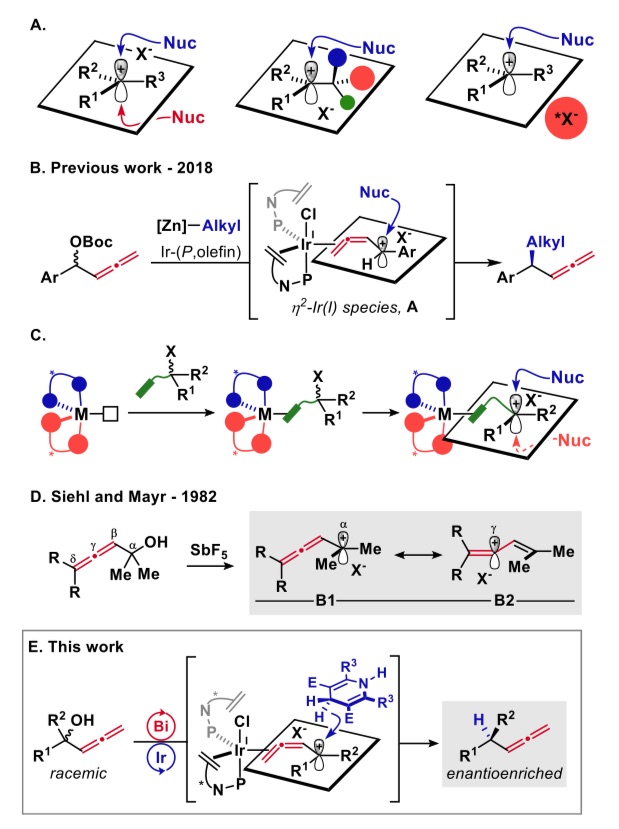
作者此次的工作即是三级碳正离子在手性环境的控制下,实现了不对称的脱氧还原反应,反应具有良好的底物适用性和优秀的区域选择性和对映选择性。
工作介绍
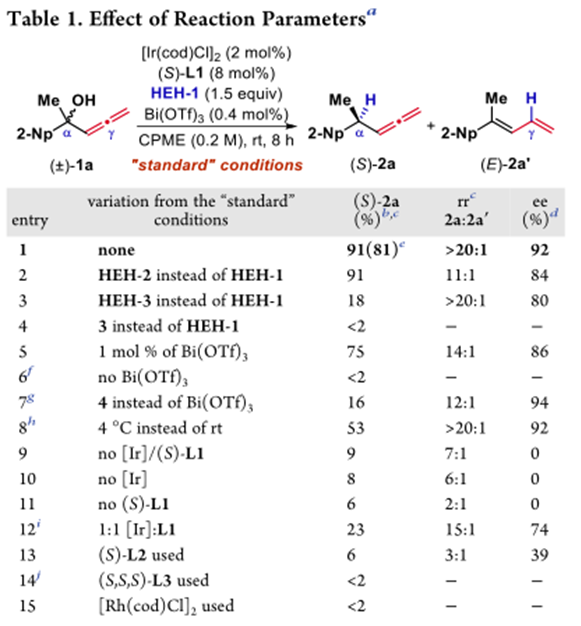
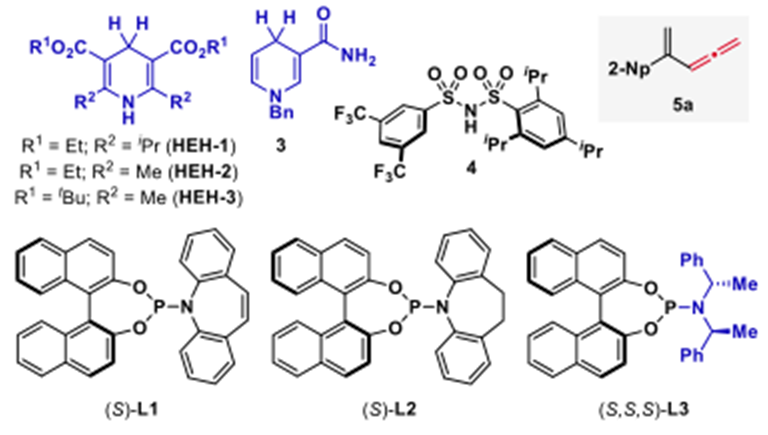
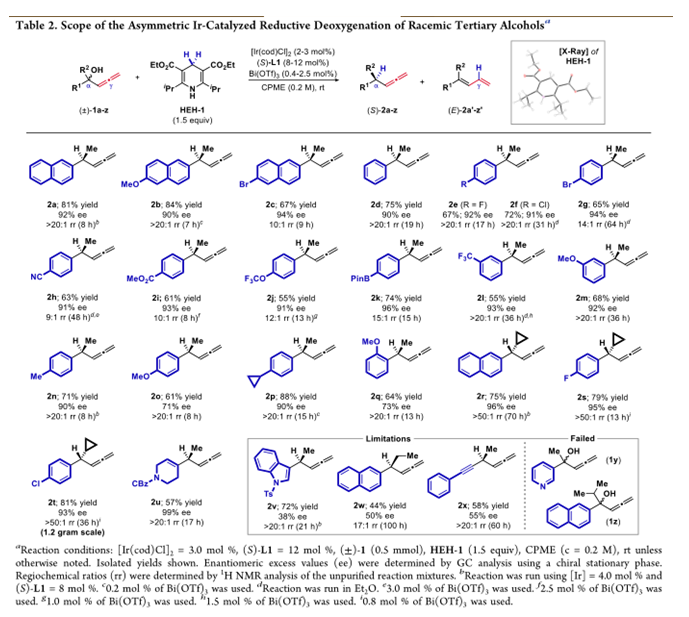
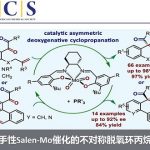
作者在考察了手性配体,Lewis酸等系列条件下,得到了最佳条件——2mol% 的[Ir(cod)Cl]2,8mol%的手性磷酸-烯烃配体,0.4mol%Lewis酸Bi(OTf)3,1.5eq还原剂HEH-1,溶剂为CPME(0.2M),室温反应。其中,Lewis酸的用量增多会使没有手性催化剂配位底物反应的背景反应增多从而使选择性下降,而使用前面反应效果良好的Bronsted酸4催化下,反应性会急剧下降,反应在低温下也可反应,选择性增加但反应性有所下降,其中Ir与配体的比例在1:2时反应有最好的反应性,而更换为Rh时,反应基本不发生。
随后,作者对反应的底物进行了拓展:当其中有一个基团为芳基时,反应对其电性有良好的兼容能力,缺电子给电子取代的底物,邻位、间位和对位都能很好兼容。当对可以稳定碳正离子的环丙基取代的反应中,都有可比的结果,但是得到的是相反的构形,并且由于环丙烷的作用,没有产生双烯产物,并且无环丙烷开环的产物生成。当使用环状内烯2u时,反应也有可比的反应结果。但是,当使用乙基,吲哚杂环和炔基时,选择性较差。另外,反应对有较高碱性的底物不适应,且当烷基位阻增大时也不反应。
由这些底物可简单得出反应趋势,富电子芳烃得到高区域选择性和较低的对映选择性,缺电子则相反。这与碳正离子的稳定作用相关。给电子和缺电子基取代芳环分别稳定α位和γ位碳正离子,当α被稳定时,形成联烯产物的比例自然会增加。
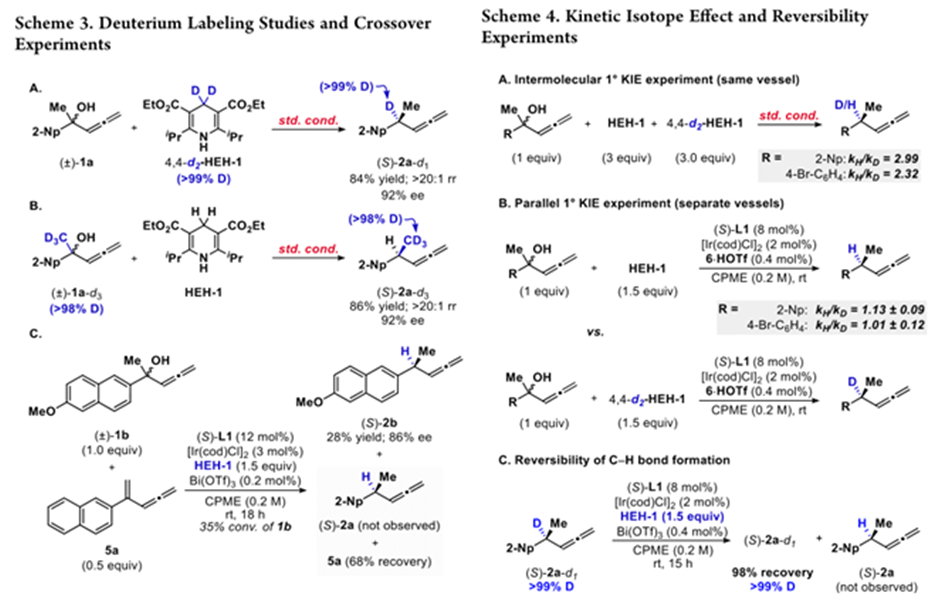
机理研究中,氘代实验表明,产物中的H全部来自还原剂。5a是反应中的一种副产物,作者对它是否为反应中间体进行氘代实验,当使用甲基氘代底物时,反应无同位素置乱产物生成,而与5a的交叉实验表明,5a并不参与到反应中,只为底物脱水后产生的副产物,且反应是通过形成碳正离子而非脱水的。
同位素反应中,当加入氘代和非氘代还原剂时,KH/KD为2.99和2.32,亲核试剂H进攻应该为决速步,但这与SN1反应机理明显不符,因此作者考虑是否氘代反应不能证明决速步[4]。而在后续分别进行的反应中(Bronsted酸代替Lewis酸),KH/KD没明显区别,佐证亲核试剂进攻并非为决速步。将氘代产物重新投入反应表明亲核进攻这一决定选择性的步骤是不可逆的。
Bi(OTf)3是一种有机溶剂中溶解性较差的无机盐,而考虑到亲核试剂进攻完全的话,反应会生成一分子带有Lewis酸性的吡啶OTf,此物种也会促进形成碳正离子的反应,因此作者做了系列对比实验验证其是否具有催化活性。在无Bi(OTf)3存在时,反应可正常进行,得到可比的结果。
随后作者做了线性自由能关系图,其拟合方程中,Hammett取代常数ρ=−1.15,表明决速步中生成了大量的正电荷,这与SN1的机理相吻合。
为了进一步验证决速步,作者做了二级同位素实验,一般来说,当从sp3→sp2转化时,由于键力常数的变化,同位素效应KH/KD为1.2-1.1,而此处同位素效应KH/KD为1.08±0.01,虽然与标准数值略有差异,但是仍然可以表明决速步中包含sp3→sp2的转化,这与SN1反应相吻合。

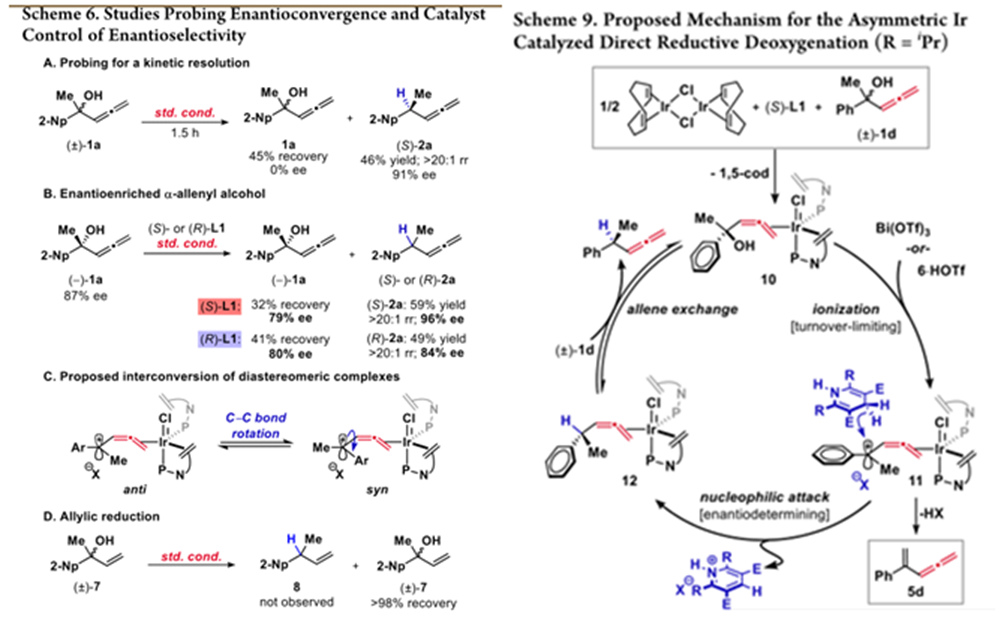
作者随后做了部分转化的实验,排除了可能存在的动力学拆分机理。而对手性底物的反应表明,底物的手性是由催化剂控制的,并且为手性控制提出一种可能的模型。最后,作者排除了烯丙基醇的反应可能性。
作者通过上述机理研究推测如下机理:配位后的底物在Lewis酸催化下形成碳正离子,随后发生C-C bondrotation后发生对映选择性的亲核进攻得到产物,最后底物产物交换重启催化循环圈。
参考文献
- Naredla, R. R.; Klumpp, D. A., Chem. Rev.,2013, 113, 6905, DOI: 10.1021/cr4001385;
- Petrone, D. A.; Isomura, M.; Franzoni, I.; Rössler, S. L.; Carreira, E. M.,J. Am. Chem. Soc.,2018, 140, 4697, DOI: 10.1021/jacs.8b01416;
- a: Siehl, H.-U.; Mayr, H., J. Am. Chem. Soc.,1982, 104, 909, DOI: 10.1021/ja00367a069; b: For a review on stable carbocations, see: Olah, G. A.; Prakash Reddy, V.; Prakash, G. K. S., Chem. Rev.,1992, 92, 69, DOI: 10.1021/cr00009a003;
- Simmons, E. M.; Hartwig, J. F., Angew. Chem., Int. Ed.,2012, 51, 3066, DOI: 10.1002/anie.201107334;
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!













No comments yet.