
导读
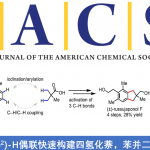
近日,上海交通大学李长坤课题组在J. Am. Chem. Soc.中报道了通过Rh与Cu在中性条件的协同催化策略,顺利实现端炔与外消旋碳酸烯丙酯之间的Sonogashira型反应,进而合成出一系列手性1,4-炔烃衍生物。该方法学具有广泛的底物适用范围,各种脂肪族及芳香族端炔化合物均能够有效地用于上述反应。作者通过对化学计量反应的研究,进而支持C(sp)-C(sp3)键生成的内球层还原消除(inner-sphere reductive elimination)机理。
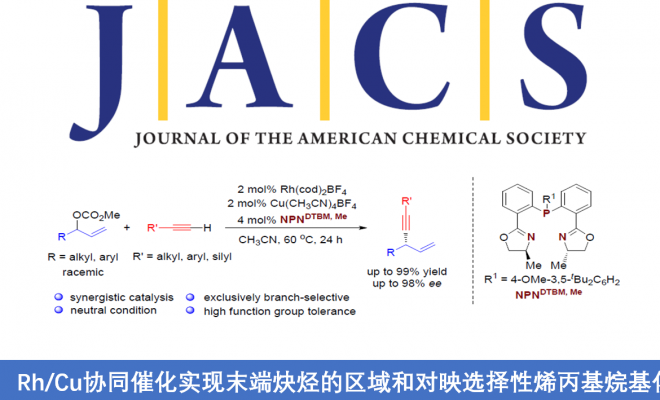
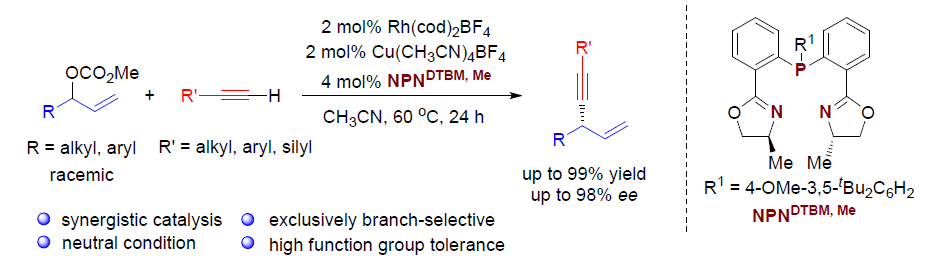
Regio- and Enantioselective Allylic Alkylation of Terminal Alkynesby Synergistic Rh/Cu Catalysis
Wen-Yu Huang, Chun-Hua Lu, Samir Ghorai, Bing Li, and Changkun Li
J. Am. Chem. Soc. ASAP. DOI: 10.1021/jacs.0c08283. https://doi.org/10.1021/jacs.0c08283
正文:
过渡金属催化的不对称烯丙基取代反应(asymmetric allylic substitution, AAS)已经成为为对映选择性地构建碳-碳键的有力方法,各类软(硬)碳亲核试剂均能够有效地参与上述反应。而端炔同样可以作为制备1,4-烯炔类化合物的潜在亲核试剂。然而,由于在常规的AAS过渡金属催化剂(如Pd、Ir与Rh)存在下,炔基化合物表现出反应活性的多样性,因此,使端炔在AAS反应中的应用受到一定限制。Hoveyda课题组报道了首例在Cu催化存在下,采用炔基铝试剂参与的不对称烯丙基炔基化反应。Carreira与Breit课题组分别通过炔基硼与炔基羧酸,成功完成高度对映体富集的1,4-烯炔化合物的合成(Scheme 1, a)。而将各种廉价易得的端炔在温和条件下直接进行不对称烯丙基化反应尽管符合原子与步骤经济性的要求,然而该研究仍存在较大的挑战。目前,文献中仅报道过两种较为成功的反应实例。Sawamura与Ohmiya课题组报道了在手性Cu催化剂存在下,端炔与(Z)-烯丙基磷酸酯之间的不对称烯丙基烷基化反应(Scheme 1, b)。研究表明,该反应条件对于三异丙基硅基乙炔或其他大位阻的炔烃,能够观察到较高的对映选择性,而芳基或脂肪族基团取代的端炔,则获得中等程度的对映选择性。Tan与Lee课题组报道了在双相条件下,铜/胍催化剂参与的环烯丙基溴的对映选择性烯丙基炔化反应。然而,在中性条件下,官能团化的端炔与廉价的非对称烯丙基前体之间的高度区域及对映选择性的直接偶联反应仍有待进一步发展。
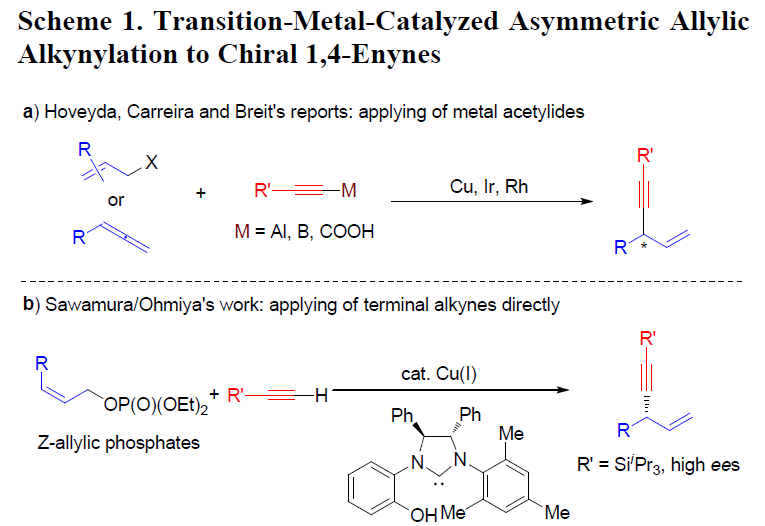
铑催化的单取代的烯丙基底物的高度支链选择性(branch-selective)的烯丙基烷基化反应已有大量文献报道。近期,本课题组发展了在中性条件下,通过Rh(I)/双噁唑啉膦催化的,各类氮、碳、以及硫前亲核试剂(pronucleophiles)参与的区域与对映选择性烯丙基化反应。该方法对于的带有烷基支链的外消旋烯丙基底物以及线性(Z/E)烯丙基底物尤其具有挑战性,这类底物在上述反应条件下,均转化为同一手性产物。反应过程中,释放出的碳酸根负离子与甲氧基负离子作为碱,使酸性的前亲核试剂发生去质子化。酸性较低的C(sp)-H键与竞争性的Rh(I)催化端炔的二聚或三聚,成为将前亲核试剂的范围扩展至端炔的主要障碍。受到Sonogashira偶联反应中Cu试剂活化端炔的启发,作者设想通过添加催化量的铜盐,能够有助于提高端炔的酸性,乙炔铜的形成以及随后向Rh中心的转金属化过程,可能会促进烯炔的产生。
在此基础上,上海交通大学李长坤课题组报道了在中性条件以及Rh与Cu协同催化下,顺利完成了具有挑战性的外消旋碳酸烯丙酯与端炔之间的高度支链选择性与高度对映选择性的烯丙基烷基化反应(Scheme 1, c)。
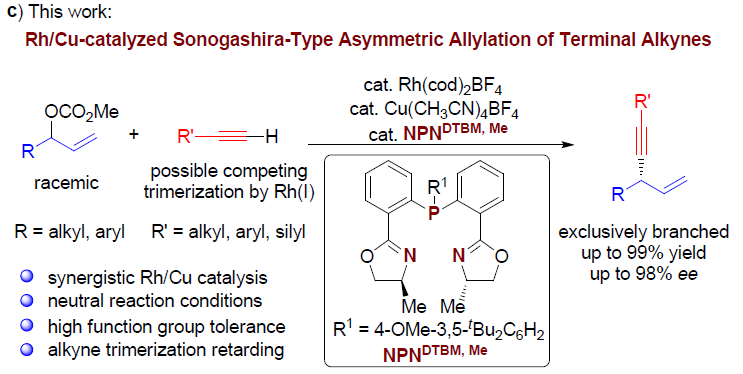
首先,作者以碳酸烯丙酯rac-1a与苯乙炔2a作为模型底物,进行了相关反应条件的筛选(Table 1)。最终筛选出最佳的反应条件为:以Rh(cod)2BF4(2 mol%)与Cu(CH3CN)4BF4 (2mol%) 作为催化剂,4 mol% L10作为配体,乙腈溶剂,反应温度为60 ℃,反应时间为24 h。

在获得上述最佳反应条件后,作者开始对末端炔烃2的底物适用范围进行系统研究(Scheme 2)。研究表明:在芳基取代端炔的芳基对位中连有供电子基与中等强度的吸电子基时,对产物3bb-3be的产率与ee几乎无显著影响。然而,芳基中连有强吸电子基(如硝基3bf)时,反应速率变慢,需将反应温度提高至80℃,收率可提升至85%,而ee略有降低。同时,芳基对位中具有甲酰基、酯基、游离磺酰胺基以及羟基(3bg至3bi,3bl)等活泼基团取代以及芳基间位与邻位的取代基(3bj至3bl),上述反应条件均能够很好地兼容。对于其它的(杂)芳环,如2-萘基、3-噻吩基以及3-吡啶基,均可以获得预期的手性产物3bm-3bo。通过三甲基硅基乙炔2p,同样能够合成出高收率与高ee的硅基取代产物3bp(进一步去保护,最终获得制备手性端炔产物)。脂肪族取代基取代的端炔同样能够应用于上述转化。游离的炔丙醇、氰基、对甲苯磺酸酯、酰胺、氯代烷、硫醚以及缩醛,在该反应条件下同样能够十分良好地兼容,进而获得相应手性产物3br-3bz。此外,1,3-烯炔底物同样能够十分容易地转化为二烯3bα与3bβ。同时,通过3bγ的高度非对映选择性合成,从而表明该方法学可以用于复杂分子的后期官能团化。而通过与文献进行比较,作者进而指认手性产物3bp与3bα的绝对构型为S-构型。
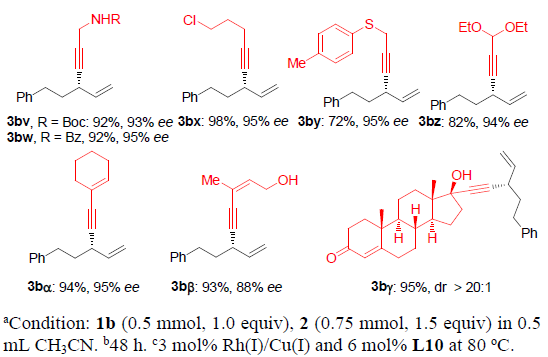
接下来,作者进一步考察烯丙基碳酸酯rac-1的底物应用范围(Scheme 3)。研究发现,在上述的标准条件下,可以顺利完成简单的甲基与正丙基取代的手性1,4-烯炔产物3cb与3db的对映选择性合成。具有较大位阻的α与β支链位置存在异丙基、环丙基、环己基以及异丁基取代的烯丙基碳酸酯同样能够顺利完成上述反应,并能够以高收率与高度对映选择性地方式获得相应的手性1,4-烯炔3eb-3hb。同时,具有挑战性的叔丁基同样能够成功地引入1,4-烯炔分子中,进而获得产物3ib,尽管ee值略有降低。.苯基取代的碳酸烯丙酯1j同样能够有效地参与上述反应,以85%的收率与97%ee获得3jb产物。通过与文献进行比较,可以确定3jb的绝对构型为R-构型。对于具有游离羟基或羟基保护基团的碳酸烯丙酯底物,能够以高ee值获得手性产物3kb与3lb。在上述标准条件下,同样可以将碳酸三级烯丙醇酯应用于上述合成转化,以中等产率与ee构建具有季碳手性中心的1,4-烯炔产物3mb-3pb。
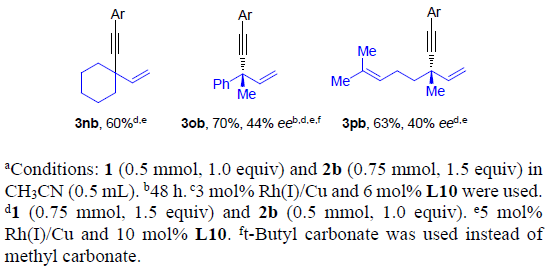
正如预期一致,Z-与E-线性的烯丙基碳酸甲酯底物(Z-4a和E-4a)能够与端炔2b进行不对称烯丙基烷基化反应,获得3db(eq 1, Scheme 4),然而,相比支链的烯丙基碳酸甲酯底物,反应速率降低。并且,要获得较高的反应收率,需要增加至3 mol%的催化剂用量以及更长的反应时间(48 h)。 同时,作者发现,上述的不对称烯丙基烷基化反应可以在10 mmol的规模下进行,仅需要1mol%催化剂,便能够以52%的收率与88%ee合成3kd(eq2, Scheme 4)。而将3kd中的羟基采用3,5-二硝基苯甲酸进行保护后,可获得酯5,并通过单晶X-射线衍射分析进一步确定其绝对构型为S-构型。

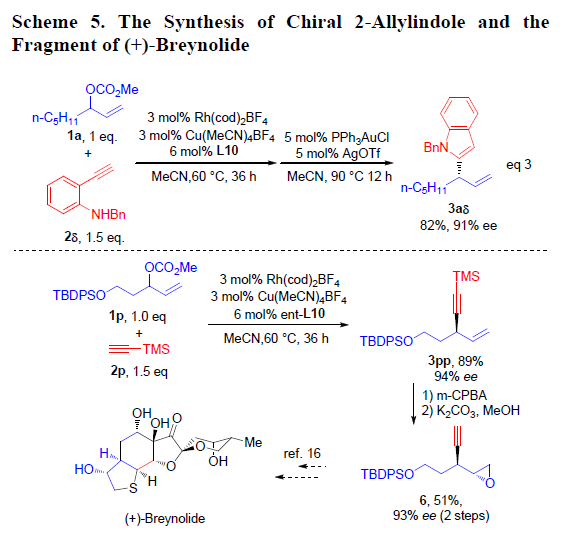
之后,作者对上述方法学在合成中的应用进行深入研究(Scheme 5)。该小组发现,通过1a与2δ在上述标准条件下完成不对称烯丙基烷基化反应后,再通过金催化的环化反应,即可获得手性N-苄基-2-烯丙基吲哚3aδ,收率为82%,ee为91%(eq 3)。此外,在ent-L10配体存在下,作者进一步将新发展的不对称烯丙基烷基化方法学应用于3pp的合成,并通过3pp作为关键砌块,进一步完成 (+)-Breynolide全合成中关键片段6的合成。
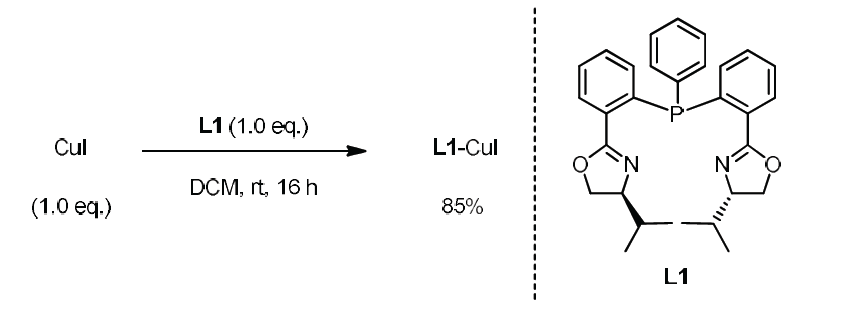
为了进一步理解Rh/Cu催化下,端炔不对称烯丙基烷基化反应的机理,作者进行了相关的控制实验与化学计量反应实验(Scheme 6)。作者观察到,在Rh/Cu与L1催化体系作用下,能够以60%的收率与90%ee分离出3ca。而在K2CO3存在下,先前报道的[L1-RhCl(MeC3H4)] OTf与乙炔铜之间的化学计量反应,未获得产物3ca。而加入1.5 eq的L1后,却能够以83%产率以及95%ee获得产物3ca。上述结果表明:加入额外的配体,对于确保较高的转化率是十分必要的。同时,加入配体L1,可能会破坏生成的乙炔铜低聚体,并产生关键中间体Cu-L1-乙炔基负离子配合物。而将CuI与L1在DCM中混合时,能够以较高收率获得具有变形四面体几何构型的配合物L1-CuI (eq 6)。此外,作者进一步观察到产物的ee%与配体之间呈现线性相关,进而表明催化过程中不涉及具有手性配体双重配位的金属配合物(详见SI)。
C-C键形成步骤不同于先前提出的氮与其他亲核试剂加成的外球层(outer-sphere addition)机理。作者根据观察到产物3bp,3bα,3jb,3ca与5的绝对构型,进而推测反应过程中更可能涉及乙炔铜向金属配合物铑中心的转金属化以及后续的还原消除途径。首先,Rh(I)/ L10与(S)-1a-Z-D之间进行立体化学反转的氧化加成过程,形成较不稳定的π-烯丙基铑配合物A。path A中,通过Rh(I)催化的烯丙基交换,迅速将配合物A转变为更稳定的配合物B。配合物B通过乙炔铜捕获后,获得主要产物(S)-3aa-Z-D。配合物B较高的稳定性可以通过分离[L1-RhCl(MeC3H4)] OTf进行证实。而path B中,通过π-σ-π异构化过程,将配合物A缓慢转化为另一个较为稳定的配合物C,进而形成次要产物(S)-3aa-E-D。主要产物(S)-3aa-Z-D的形成支持内球层机理(inner-sphere mechanism)。
总结
上海交通大学李长坤课题组报道了通过中性条件以及Rh与Cu催化剂的协同催化下,端炔与外消旋碳酸烯丙酯之间的不对称烯丙基烷基化方法学,进而合成出一系列的手性1,4-炔烃衍生物。该反应具有广泛的底物应用范围以及优良的官能团兼容性。通过加入乙腈溶剂,能够抑制Rh(I)催化的炔三聚副反应。而化学计量反应与控制实验的研究支持C-C生成过程中涉及内球层(inner-sphere)还原消除的机理。

















No comments yet.