
本文作者:杉杉
导读
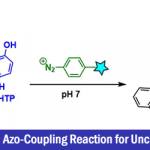
N-芳基唑骨架(Aryl azole)广泛存在各类药物分子中,由于酸性的杂环具有竞争性的金属化作用(metalation),导致芳环上的邻位选择性金属化极具挑战。为了开发一种关键活性药物成分(API)中间体的实用途径,作者研究了具有空间位阻的金属酰胺碱(metal-amide bases)与1-芳基-1H-1,2,3-三唑和其它杂环的金属化作用。在此,德国慕尼黑大学Paul Knochel教授与瑞士杨森制药公司(Janssen Pharmaceutica)Simon Wagschal共同在Nature Communications上报道了,可在室温条件下,烃类溶剂中使用氨基化镁(magnesium amide,TMPMgBu(TMP = 2,2,6,6-四甲基哌啶基),实现对几种芳基唑的高度区域选择性的邻位金属化,然后有效进行了Pd催化的芳基化反应。这种可扩展和选择性的反应允许改变芳基环的取代模式,唑部分的性质以及亲电试剂的性质。这种通用的方法可以应用于生物活性唑衍生物的合成,并补充现有的金属介导的邻位官能化反应。
Regioselective functionalization of aryl azoles aspowerful tool for the synthesis of pharmaceutically relevant targets
Ferdinand H. Lutter, Lucie Grokenberger, Luca Alessandro Perego , Diego Broggini,
SébastienLemaire, Simon Wagschal& Paul Knochel
Nat. Commu. ASAP DOI:10.1038/s41467-020-18188-z
前言
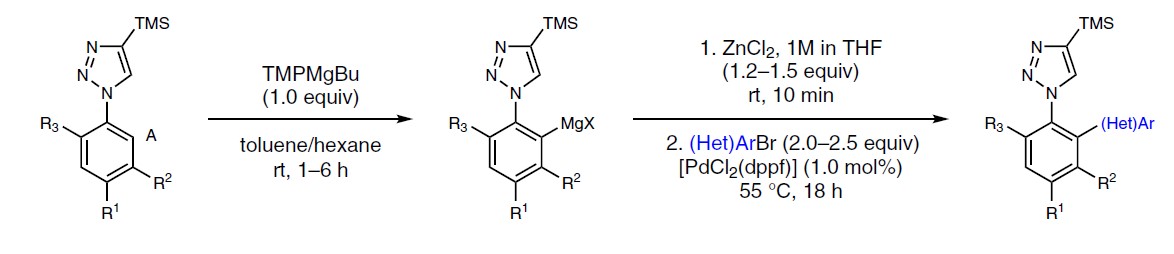
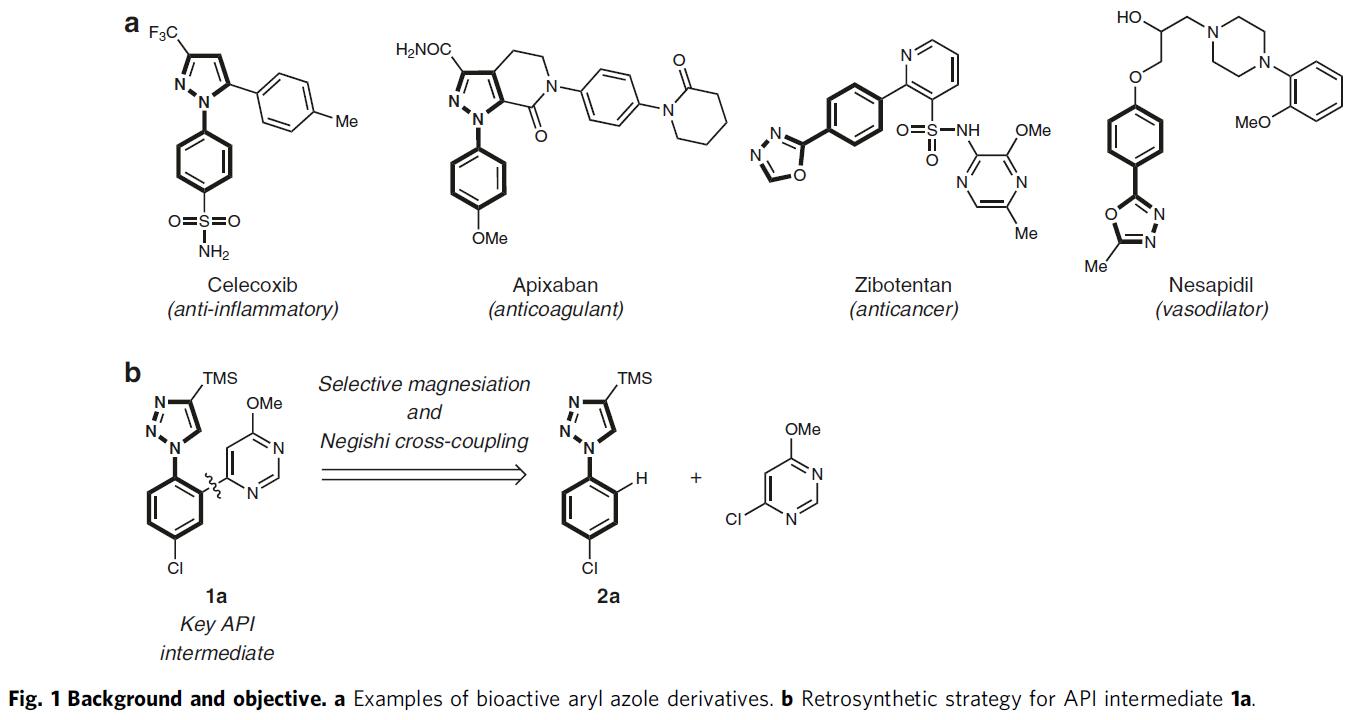
N-芳基唑骨架存在于多种市售的药物中,如塞来昔布(celecoxib)、阿哌沙班(apixaban)、齐泊腾坦(zibotentan)和奈沙地尔(nesapidil)(Fig. 1a)。若直接实现1-芳基-1H-1,2,3-三唑(2a)的邻位C-H功能化,则可直接获目标产物1a(Fig. 1b)。对于涉及过渡金属催化的C-H芳基化的方法,常需苛刻的条件,并产生双芳基化产物,从而限制了它们的实用性。因此,使用合适的碱直接去质子化,是芳基唑选择性官能化的替代方案。然而,由于N-杂环本身具有竞争性的金属化作用,与杂环相连的芳基环的区域选择性金属化极具挑战。
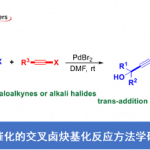
在芳环上实现区域选择性金属化的潜在方法中,由于溶剂中存在供电子效应,与吡咯环的氮原子存在竞争,需避免配位溶剂(如THF)。立体受阻的金属酰胺碱(metal-amide bases)中,尤其是镁和锌衍生的TMP碱作为各种(杂)芳烃官能化的有效试剂,越来越被重视。Hagadorn报道,TMP2Zn是各种羰基化合物的α-锌离子化反应和吡啶-N-氧化物在甲苯中金属化反应的最佳碱。同样,Mulvey课题组报道了几种在非配位烃溶剂中,用于金属化反应的混合双金属酰胺碱。在此,德国慕尼黑大学Paul Knochel教授与瑞士杨森制药公司Simon Wagschal共同报道了在甲苯/己烷溶剂混合溶剂中使用酰胺碱TMPMgBu,各种芳基唑的可以进行高度选择性和广泛适用性的交叉偶联和亲电淬灭反应。
反应条件的优化
作者以1-芳基-1H-1,2,3-三唑2a与各种金属酰胺碱的反应为模板,以评估产物A和B之间的选择性(Fig. 2a)。反应结果表明,当以TMPMgBu作为碱金属(通过使用市售的Bu2Mg的己烷溶液与TMP-H反应制备),并在甲苯溶剂中反应,可在1h内以81%的收率获得所需的金属化三唑产物A(A:B = 96:4,entry 7)。随后,使用ZnCl2转金属化(transmetalation),再使用1 mol%的PdCl2(dppf)将所得的芳基锌试剂与4-氯-6-甲氧基嘧啶偶联,以86%的收率获得所需的活性药物成分(API)中间体1a(Fig. 2c)。

底物范围的扩展
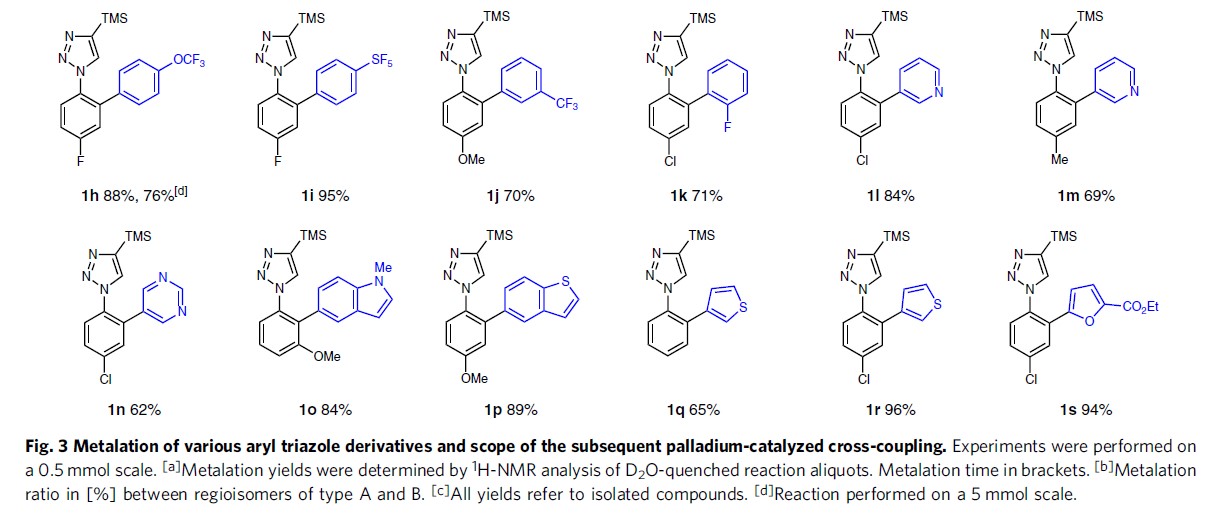
在获得上述最佳反应条件后,作者研究各种取代的芳基三唑的金属化反应的范围(Fig. 3)。缺电子2b、未取代2c、富含电子2d–2f以及邻氟2g的底物均可获得相应的有机镁试剂3b–3g。值得注意的是,富含电子的底物金属化的时间较长。同时,并未观察到甲氧基或氟部分的邻位金属化,表明三唑单元作为较强的导向基团。
在获得上述的金属化试剂后,作者开始进行下一步的偶联反应。具有富电子或缺电子的芳基溴底物均可顺利进行,以75-87%的收率获得相应的产物1b–1f。具有空间位阻的2-溴萘也以74%的收率获得产物1g。含有三氟甲氧基、五氟亚磺酰基、三氟甲基或氟取代基的各种氟化芳基溴化物也适用于该偶联反应,以70-95%的收率获得所需的芳基化产物1h–1k。此外,一系列杂芳基溴化物底物,如吡啶基、嘧啶基、吲哚基、噻吩基、呋喃基等溴化物同样可以作为偶联底物,获得相应产物1l–1s,产率为62-96%。

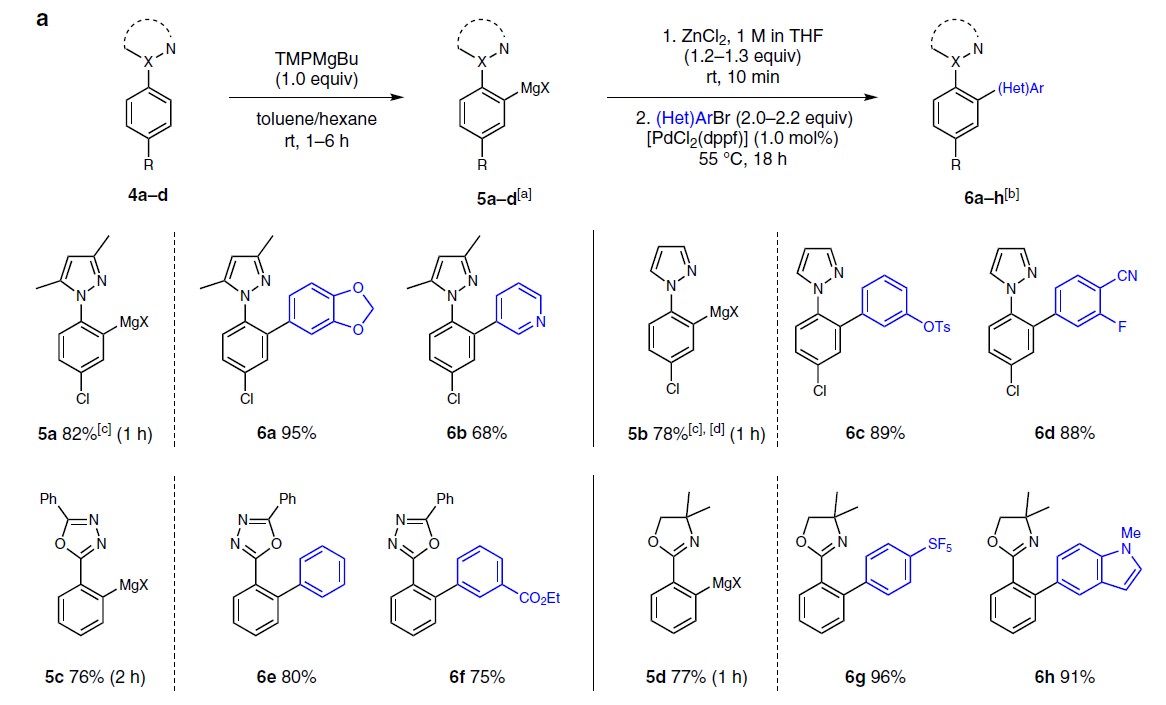
接下来,作者将金属化作用扩展到其它芳基唑的底物(Fig. 4a)。使用TMPMgBu与1-苯基-3,5-二甲基-1H-吡唑4a 反应1h,获得82%收率的5a。使用未取代的吡唑4b,获得78%收率的5b。值得注意的是,在任何情况下均未观察到竞争性的唑环金属化。此外,对2,5-二苯基-1,3,4-恶二唑4c可在2 h金属化后以76%的收率得到5c。苯基恶唑啉4d可在1 h内,以77%的收率得到5d。
随后,作者将5a–5d在标准条件下进行下一步的偶联反应,以68-95%的收率获得产物6a–6h。值得注意的是,含有3,5-二甲基吡唑基的底物5a,可通过臭氧的氧化裂解获得相应的N-乙酰化苯胺。同时,合成的6e可作为电致发光化合物的宝贵前体。
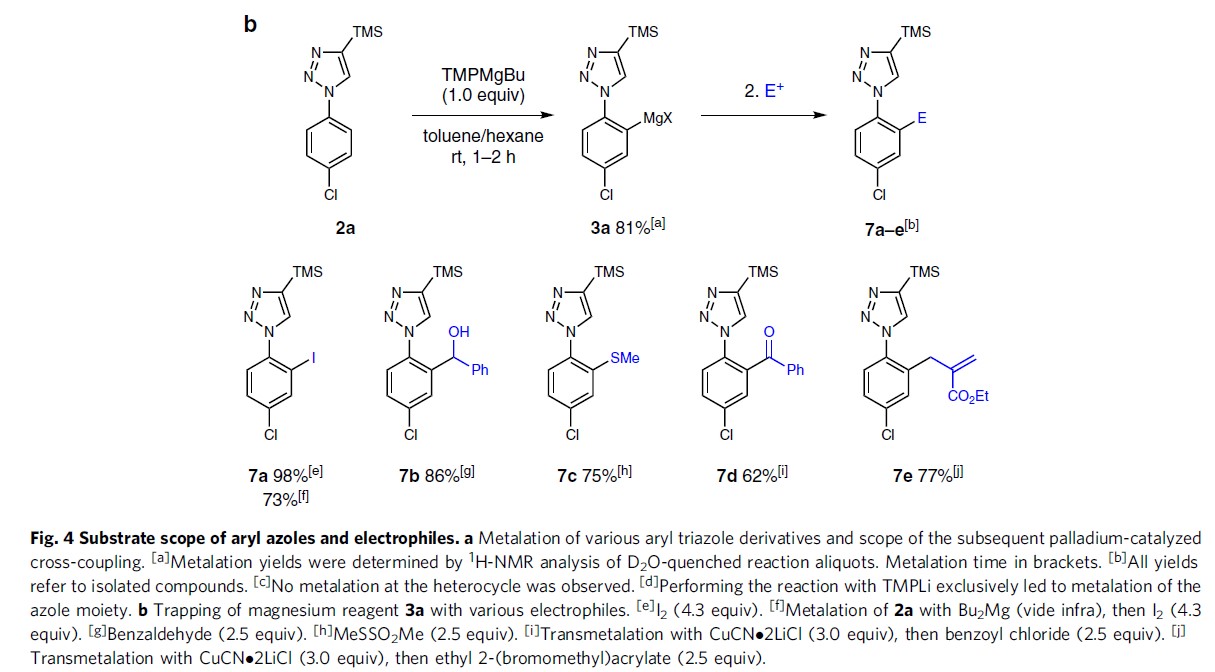
此外,作者还将芳基镁试剂3a与其它常见的亲电试剂反应(Fig. 4b)。若与I2反应获得98%收率的7a。若与苯甲醛或MeSSO2Me,分别以86%和75%的产率得到相应的醇7b或硫醚7c。若用CuCN⋅2LiCl进行金属转移,然后与苯甲酰氯或烯丙基溴衍生物反应,得到7d–7e,产率为62-77%。
后期修饰
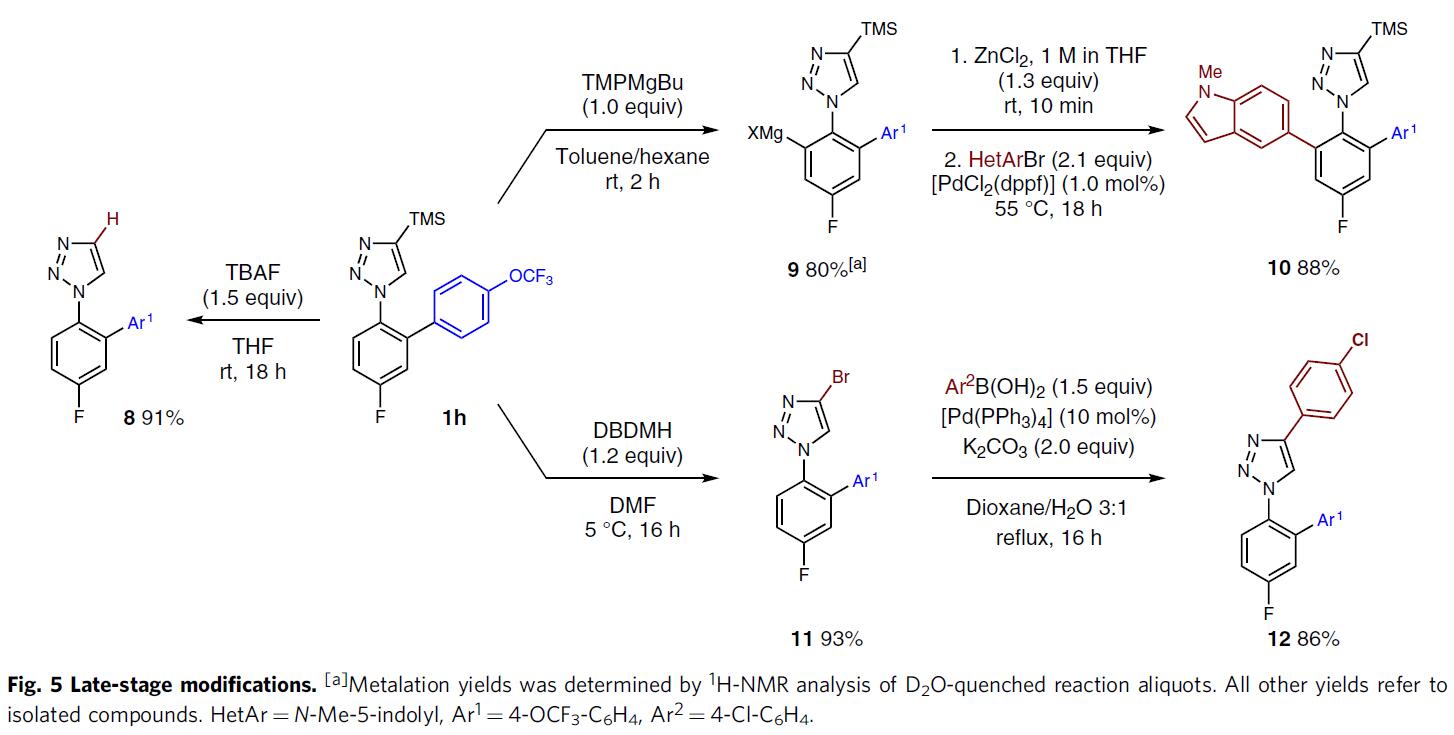
为了进一步证明反应的实用性,作者进行了相关的后期修饰(Fig. 5)。若使用TBAF可以轻松除去TMS基团,从而以91%的产率获得未取代的三唑8。若1h与TMPMgBu在甲苯中反应2小时,可得到芳基镁试剂9(产率为80%),再在标准的偶联条件下反应,可获得88%产率的双芳基化的三唑10。若1h与1,3-二溴-5,5-二甲基乙内酰脲反应,以93%的产率获得相应的溴化物11,可进一步与苯硼酸进行Suzuki交叉偶联,从而以86%的收率得到12。
反应机理
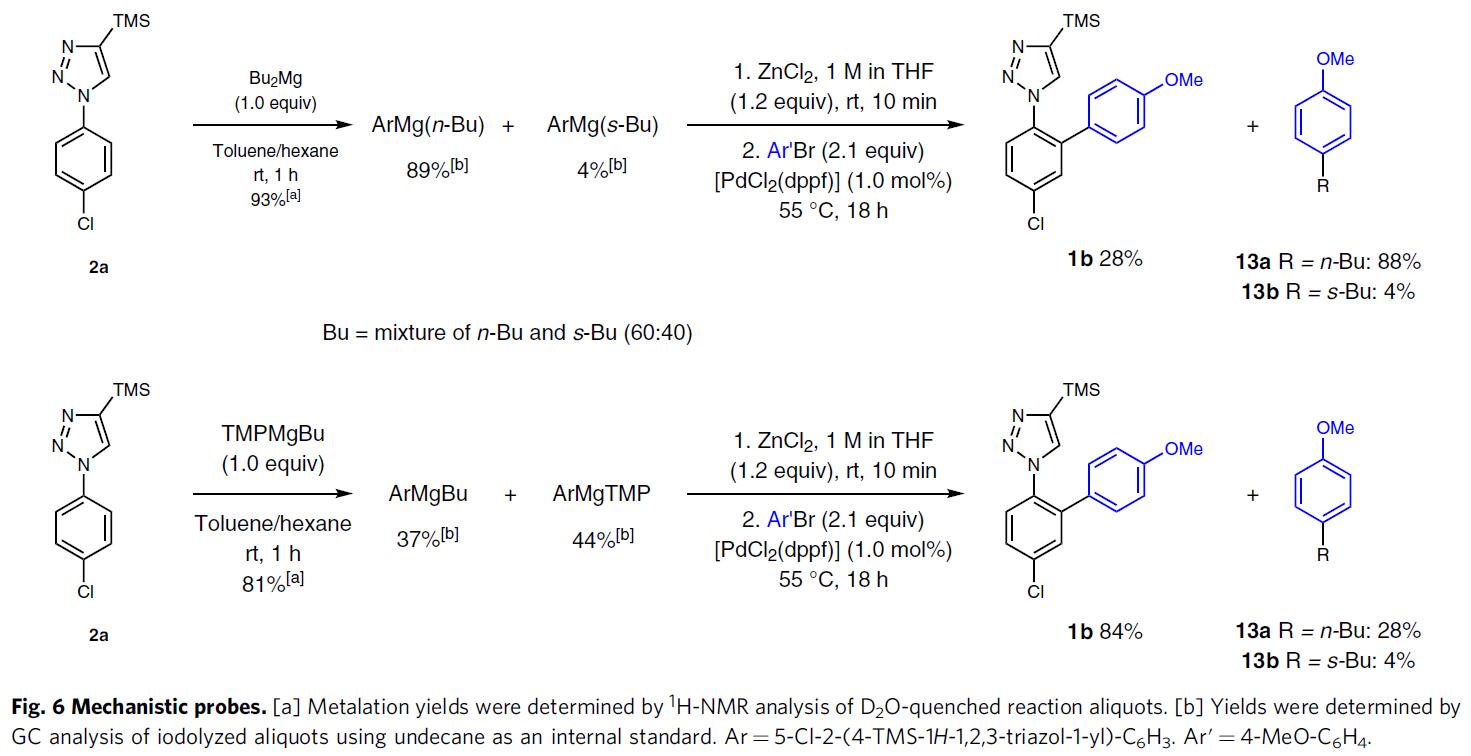
为了进一步了解反应的机理,作者进行了相关的对照实验(Fig. 6)。已知市售的Bu2Mg溶液(正丁基和仲丁基的比例为6:4),而在TMPMgBu中的两者比例相同。当在甲苯/己烷中,Bu2Mg也是选择性脱质子化2a的极好碱(93%产率),所得混合物主要包含ArMg(n-Bu)和ArMg(s-Bu)(分别为89%和4%)。然而,在用氯化锌进行金属转移后,由于正丁基的交叉偶联,仅获得了28%的所需交叉偶联产物1b和88%的4-丁基茴香醚(13a)。该结果说明,在金属化/交叉偶联的过程中,TMPMgBu优于Bu2Mg,TMPMgBu的使用限制了ArMgBu的形成,因此在ArZnBu重金属化后优先将丁基转移至Ar’Br,形成副产物Ar’Bu(13a)。
总结
本文主要描述了,可在室温条件下,在烃类溶剂中使用氨基化镁(TMPMgBu),可实现对几种芳基唑的高度区域选择性的邻位金属化,然后在钯催化可与多种(杂)芳基溴化物或亲电试剂进行芳基化反应。同时,该方法学可以应用于关键API中间体的合成,并且产物的后期修饰进一步证明了反应的实用性。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!












No comments yet.