
本文作者:ChemBoy
导读
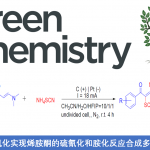
最近,西班牙加泰罗尼亚化学研究所的Ruben Martin课题组成功利用第一例[1,4]-Ni迁移过程,实现了远程芳烃C(sp2)-H键与CO2的羧基化反应。该反应作为第一例催化[1,4]-Ni迁移反应,为镍催化还原性交叉偶联反应的领域开辟了一条新的路径。同时,也为远程C(sp2)-H键上引入亲电体提供一个新的平台。该研究工作发表在JACS上:
“Remote sp2 C−H Carboxylation via Catalytic 1,4-Ni Migration with CO2”
Marino Borjesson, Daniel Janssen-Muller, Basudev Sahoo, Yaya Duan, Xueqiang Wang, and Ruben Martin*
J. Am. Chem. Soc. 2020, 142, 16234. DOI: 10.1021/jacs.0c08810
前言
近年来,已报道多种关于芳基(拟)卤化物与CO2的催化还原性羧基化合成苯甲酸反应的设计与发展,其中苯甲酸作为生物活性分子中的重要部分。尽管该领域已经到达了较高的水平,但是在CO2插入前需要对目标C(sp2)反应位点进行预官能团化(Scheme 1, path a)[1]。毫无疑问,寻求一种催化的C(sp2)-H键直接羧基化的方法(Scheme 1, path b)[2],除了在概念和实用性方面,对化学开发都是有价值的努力。
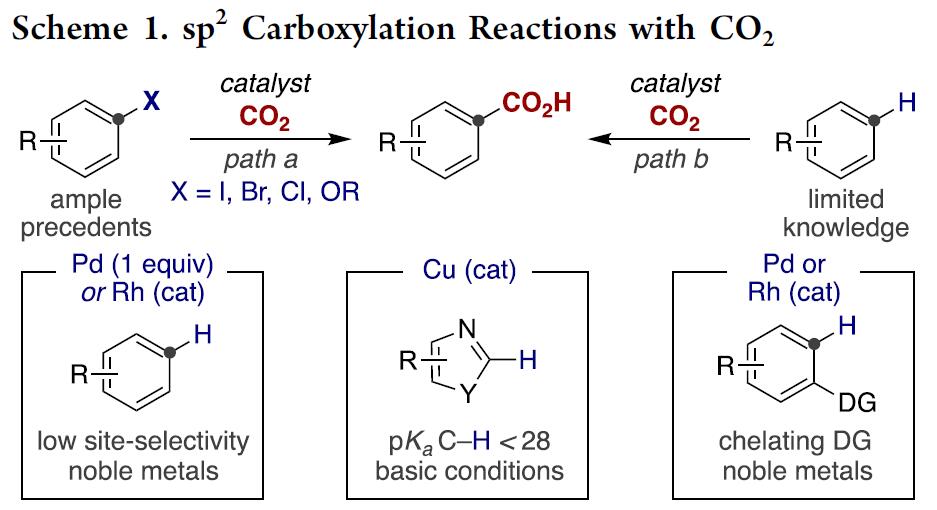
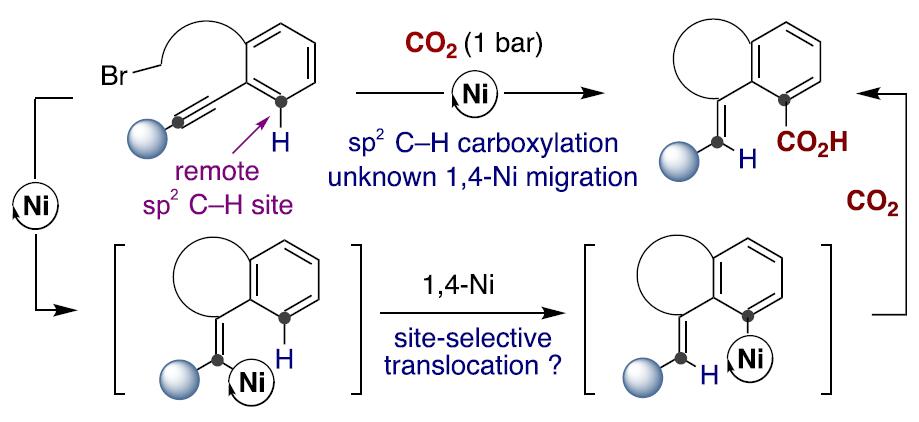
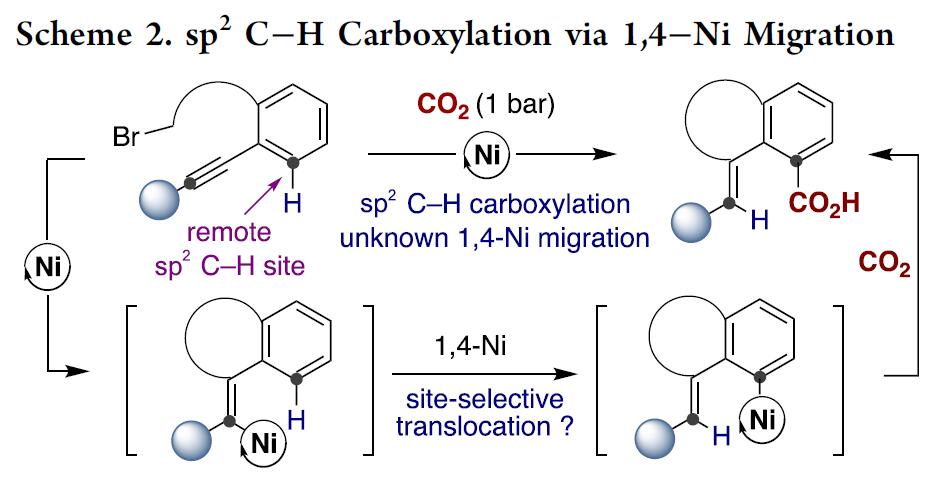
受Fujiwara报道的化学计量Pd配合物这一开创性工作的启发[3],出现了一些重要的催化酸性C(sp2)-H键[4]或导向基导向的C(sp2)-H键直接羧基化反应方法的报道(Scheme 1, bottom)[5]。然而,对于非酸性C(sp2)-H键的直接羧基化,在没有导向基团存在的情况下,会出现位置选择性问题,并且需要使用贵金属或化学计量的有机金属试剂(Scheme 1, bottom left)[6]。这些结果让有机化学家们意识到,无需贵金属或有机金属试剂进行的催化C(sp2)-H羧基化策略可能会为C(sp2)-H键官能团化与羧基化提供新的基础知识。受此启发,作者设想是否可通过基于位置选择性的[1,4]-Ni迁移[7]的串联过程实现CO2插入远程C(sp2)-H键的反应(Scheme 2)。作者在开始研究之前,这种策略能否成功未知,因为有以下两点原因:(1)目前,在交叉偶联反应中未出现催化的[1,4]-Ni迁移过程的报道;(2)在反应中由于C(sp3)-Br键与炔基活泼反应位点的存在,会遇到位置选择性的问题[8]。如果该设想能够成功,这将成为还原性交叉偶联反应领域的重大突破,可实现其他方法不能实现的C(sp2)-H键功能化的反应,从而作为一种新型反应模式。
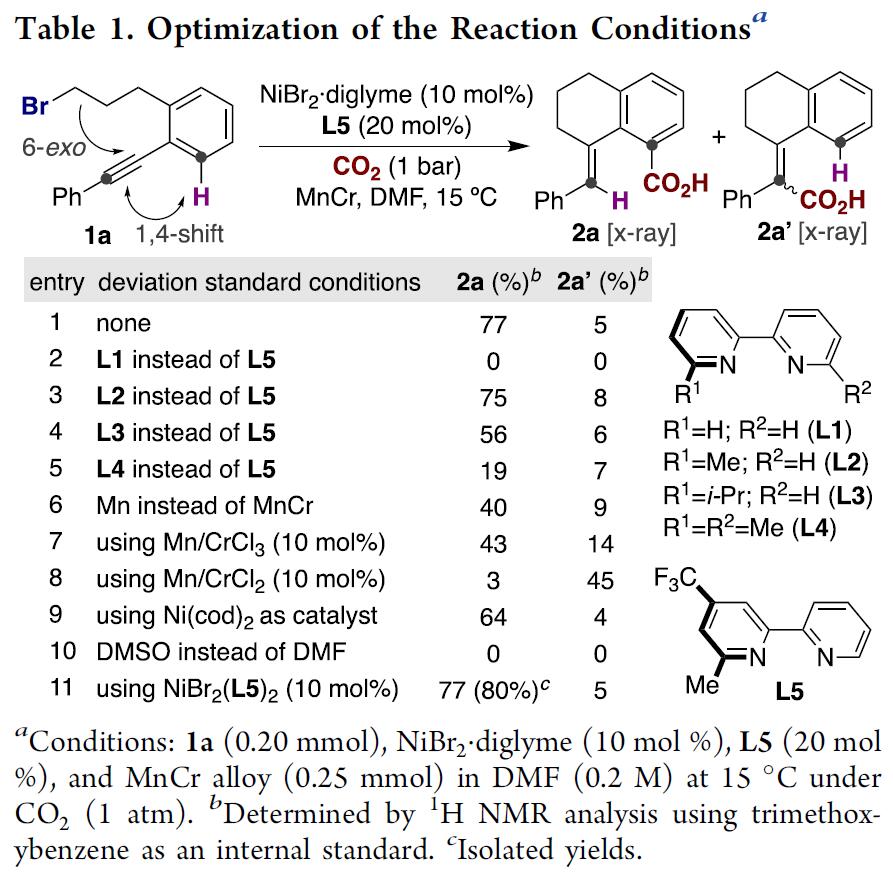
反应条件优化
首先,作者以化合物1a作为模板底物、以CO2为羧基源对反应条件进行筛选(Table 1)。不出所料,当使用以前报道的芳基卤化物与CO2的还原性羧基化反应条件时[9],仅观察到微量的目标产物2a,主要是生成了CO2与炔基反应的副产物2a’。在对反应参数进行优化后,发现NiBr2.diglyme、L5与Mn89Cr11的组合使用是反应成功的关键,能以77%的产率与优秀的位置选择性(2a:2a’=94:6)生成目标产物2a(Table 1, entry 1)。随后,作者对配体核心骨架2,2-联吡啶的电性或位阻进行调整,发现对反应活性有不可忽视的影响(Table 1, entries 2-5);显著地,使用其他类金属还原剂替代Mn89Cr11时,反应的产率与选择性均明显降低(Table1, entries 6-8);尽管有人认为Cr原子的存在可能决定了反应的位置选择性,而entry 7与entry 8证明结果却相反,目前作者对Cr的作用也无法解释。接着作者发现使用Ni(cod)2作为催化剂或DMSO替换DMF作为溶剂时(entry 9 and entry 10),均无法达到理想的结果。然而,当使用对空气与湿气稳定的NiBr2(L5)2作为催化剂时,能以优秀的产率与位置选择性生成目标产物,作者以此为最优反应条件(Table 1, entry 11)。
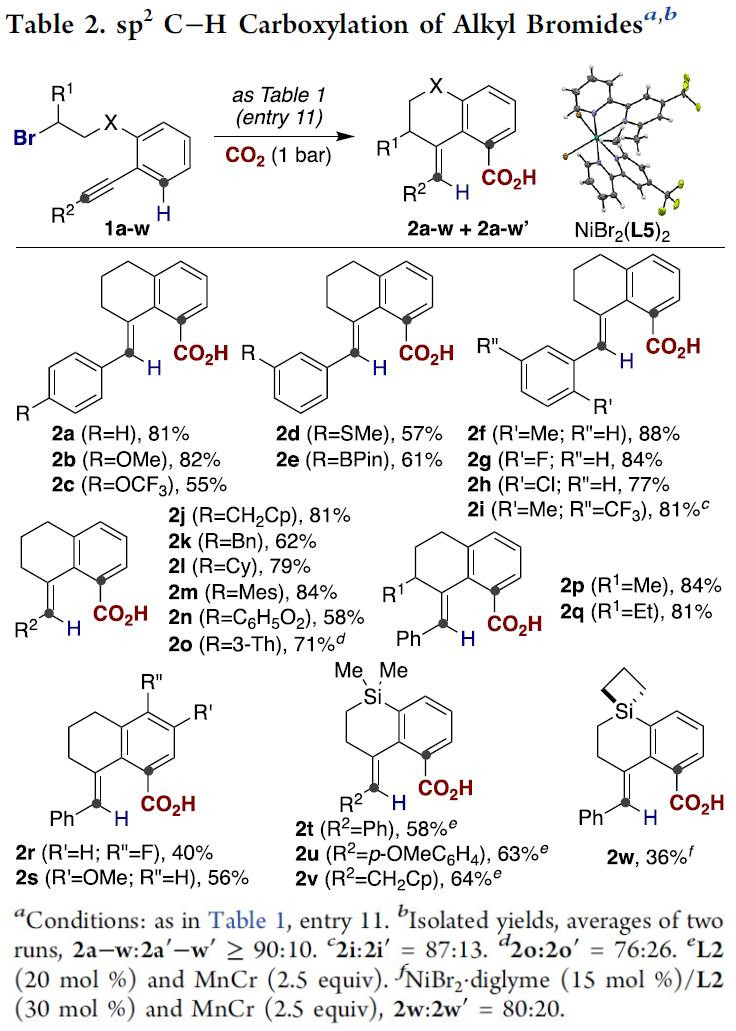
底物拓展
在确定最优反应条件后,作者对该反应的底物范围进行探索(Table 2)。作者发现,底物结构中含有硫醚(2d)、甲氧芳烃(2b)、三氟甲基衍生物(2c and 2i)或杂环(2o)时,反应均能完美地进行。甚至芳基卤化物(2g, 2h and 2r)或有机硼化物(2e)也能兼容。值得注意的是,炔烃部分无论是大位阻芳基(2m)或脂肪烷基取代(2j-l)底物也能以优秀的产率获得目标产物。无论是炔烃骨架上的位阻因素还是惰性二级烷基溴化物(2p and 2q)均能以较高的产率生成目标C(sp2)-H键羧基化产物。有趣的是,芳环上存在不同取代基时也不能阻止[1,4]-Ni迁移过程的发生,尽管反应的产率比较低(2r and 2s);此外,通过Si原子链接烷基溴化物尤其重要,也能获得目标羧酸产物(2t-w),所得羧酸产物可通过后期的C-Si键断裂生成非稠环类化合物(Scheme 4, bottom)。
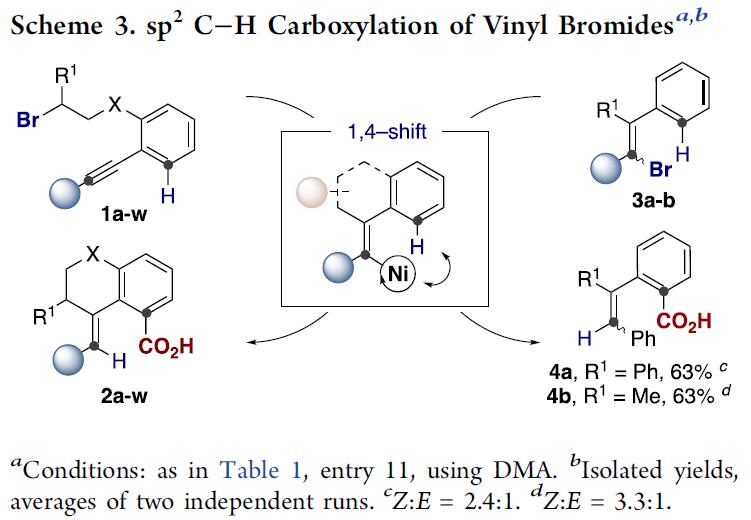
化合物2a-w的成功合成,作者认为简单烯基卤化物作为底物时应该也能发生类似的[1,4]-Ni迁移的远程C(sp2)-H键羧基化反应。如Scheme 3所示,作者将反应溶剂由DMF改为DMA后,其他条件不变,发现无论是烷基取代还是芳基取代的烯基溴化物均能发生目标C(sp2)-H键直接羧基化生成目标产物4a(E/Z=2.4/1)与4b(E/Z=3.3/1), 尽管底物3a与3b均为E/Z异构体混合物。实验结果表明,底物与催化Ni(0)物种发生氧化加成形成的Ni(II)物种在发生[1,4]-Ni迁移步骤之前会发生E/Z异构化,过程中可能存在卡宾类中间体[10]。
衍生化研究
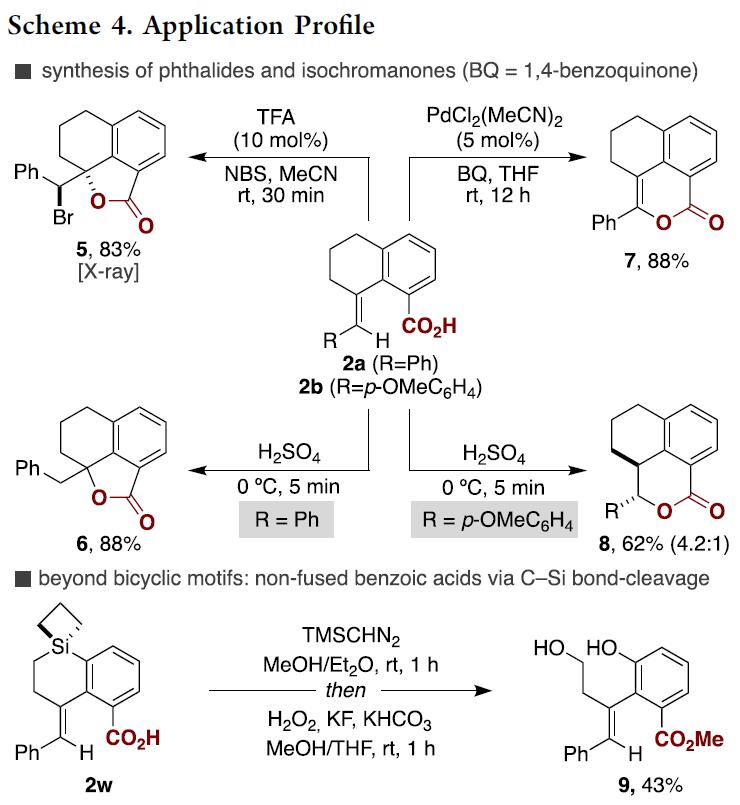
此外,作者为了证明[1,4]-Ni迁移技术的应用价值,对羧基化产物进行了一系列的衍生化应用(Scheme 4)。羧酸2a或2b在两种不同的反应条件下能够合成异色满酮或苯酞类复杂分子,羧酸2a在TFA( 10mol%)、NBS存在条件下发生溴内酯化以83%的产率生成苯酞类化合物5,而在H2SO4作用条件下能以88%的产率生成苯酞类化合物6;相似地,2a在PdCl(MeCN)2催化作用下能以88%的产率获得异色满酮类化合物7,同时2b在H2SO4存在条件下能以62%的产率获得异色满酮类似物8(dr 4.2:1)。重要的是,存在Si原子的羧酸2w 在TMSCHN2、H2O2作用下能够发生C-Si键断裂以43%的产率获得多羟基羧酸酯类化合物9,进一步证实了该羧基化反应方法的实用性。
反应机理探索
最后,作者对反应机理进行了初步研究(Scheme 5)。首先,作者以3c作为底物进行了同位素标记研究(Scheme 5, top),3c在最佳反应条件下进行反应以45%的产率生成目标产物4c,其中4c的烯基位的氘代率大于95%,证明在CO2插入目标C(sp2)-H键前在烯基邻位C(sp2)-H键上发生了[1,4]-Ni迁移;有趣的是,对化合物3d进行还原性羧基化反应时,发现了明显的分子内动力学同位素效应(KH/KD=5:1; Scheme 5, top right),作者通过3a与3c的反应速率对比未发现分子间动力学同位素效应。这些结果表明,在发生[1,4]-Ni迁移前或迁移后存在一个决速步骤。尽管如此,仍不知CO2插入与[1,4]-Ni迁移过程是发生在Ni(I)还是Ni(II)金属中心上。因而,作者将注意力转移到氧化加成形成的烯基或芳基镍金属配合物中间的分离上(Scheme 5, bottom)。有趣的是,芳基(烯基)卤化物与Ni(cod)2和TMEDA或L2在THF溶剂中能发生反应分别生成镍有机金属络合物10与11。作者以这两种镍络合物作为反应物进行羧基化反应研究,发现10仅在L5存在条件下就能与CO2发生反应以49%的产率生成目标羧基化产物4a, 证实2,2’-联吡啶配体对发生目标羧基化反应的重要性;更重要的是,10与11均能在无外加金属还原剂存在条件下发生羧基化反应生成目标产物4a, 进一步证明[1,4]-迁移过程的存在和CO2的插入发生在Ni(II)金属中心上。我们不能低估这一实验结果,因为它挑战了之前普遍的观点,即CO2的插入与[1,4]-迁移发生在Ni(I)金属中心上。从而为催化剂的设计提供了一种新的思路,尤其是在还原性交叉偶联反应领域。
总结与评价
Martin教授课题组报道了首例以催化的1,4-Ni迁移作为工具促使CO2与远程C(sp2)-H键的直接羧基化反应方法,该策略无论是在羧基化还是还原性交叉偶联领域均未被其他课题组报道过,并且该方法具有精准的化学与位置选择性、反应条件温和与实用性的特点。
参考文献
- [1] Tortajada, A.; Julia-Hernandez, F.; Borjesson, M.; Moragas, T.; Martin, R. Angew. Chem., Int. Ed. 2018, 57, 15948.DOI: 10.1002/anie.201803186
- [2] Hong, J.; Li, M.; Zhang, J.; Sun, B.; Mo, F. ChemSusChem 2019, 12, 6. DOI: 10.1002/cssc.201802012
- [3] Sugimoto, H.; Kawata, I.; Taniguchi, H.; Fujiwara, Y. J. Organomet. Chem. 1984, 266, C44. DOI:10.1016/0022-328X(84)80150-3
- [4] Zhang, L.; Cheng, J.; Ohishi, T.; Hou, Z. Angew. Chem., Int. Ed. 2010, 49, 8670. DOI:10.1002/anie.201003995
- [5] Mizuno, H.; Takaya, J.; Iwasawa, N. J. Am. Chem. Soc. 2011, 133, 1251.DOI: 10.1021/ja109097z
- [6] Suga, T.; Saitou, T.; Takaya, J.; Iwasawa, N. Chem. Sci. 2017, 8, 1454.DOI: 10.1039/c6sc03838g
- [7] Ma, S.; Gu, Z. Angew. Chem., Int. Ed. 2005, 44, 7512. DOI: 10.1002/anie.200501298
- [8] (a) Julia-Hernandez, F.; Moragas, T.; Cornella, J.; Martin, R. Nature 2017, 545, 84.DOI: 10.1038/nature22316. (b) Gaydou, M.; Moragas, T.; Julia-Hernandez, F.; Martin, R. J. Am. Chem. Soc. 2017, 139, 12161.DOI: 10.1021/jacs.7b07637
- [9] Fujihara, T.; Nogi, K.; Xu, T.; Terao, J.; Tsuji, Y. J. Am. Chem. Soc. 2012, 134, 9106. DOI: 10.1021/ja303514b
- [10] Liu, X.; Li, X.; Chen, Y.; Hu, Y.; Kishi, Y. J. Am. Chem. Soc. 2012, 134, 6136. DOI:10.1021/ja302177z
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
















No comments yet.