
投稿作者 陈元金
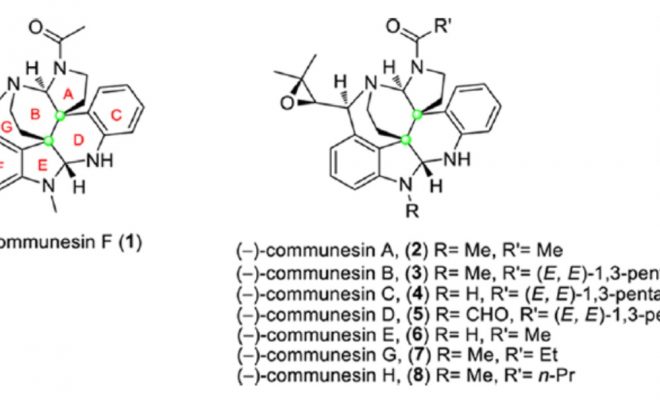
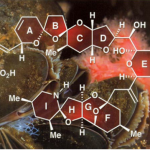
吲哚生物碱 Communesin 家族是 8 个具有复杂七环系的天然产物。因具有独特的化学结 构(4 个氮原子构成罕见的上下两个 aminal 缩醛胺基团,外加两个连续手性季碳)和有趣的生物活 性而成为明星分子,尤其是 Communesin F 的全合成备受化学家青睐。完成 Communesin 这 样高难度的分子需要设计巧妙、新颖的合成策略。基于对现有 5 条全合成路线的分析,杨玉 荣等人发现目前尚未有效解决两个关键问题:(1)Communesin F 的催化不对称的全合成;(2) 下方 aminal 基团的四环体系一步仿生构建。为此,研究人员创新性设计出 Ir 催化新策略, 实现了 3-取代吲哚和苯胺衍生的烯丙基亲电试剂,分子间的一步仿生环化,高效构建 CDEF 四环。该串联反应具有高度选择性(对映体、非对映体,区域)和经济性(步骤、原子、氧 化还原)的优点。在全合成最后阶段,利用扭曲酰胺 LAH 还原为 O, N-hemiaminal,甲磺酰 化伴随亚胺离子的环化反应得到复杂七环骨架,脱 Boc,简洁完成了(-)-communesin F 的催 化不对称全合成。 [1]这一工作十分漂亮,最为突出的是 Tetracyclic Aminal 的构筑和最后一步 的还原关环反应。下面就这一工作进行详细解读。
Ir-Catalyzed Asymmetric Total Synthesis of (−)-Communesin F
Xiao Liang, Tian-Yuan Zhang, Xue-Yi Zeng, Yu Zheng, Kun Wei, and Yu-Rong Yang.
J. Am. Chem. Soc.2017, 139, 3364−3367. DOI: 10.1021/jacs.7b00854
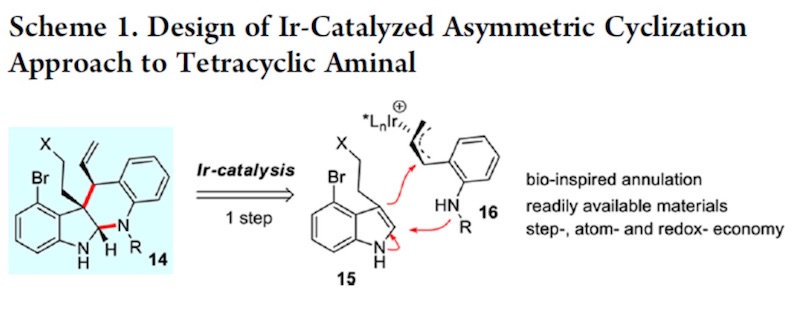
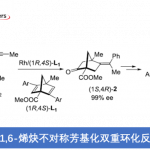
首先作者对四环体系构筑的方法学进行研究。作者根据之前自己课题组的工作,试图用 铱催化不对称环化的方法进行构筑。如 scheme 1.
于是,作者用之前已报道过的条件,对 17,18 进行的反应进行研究。遗憾的是,并未得到目标化合物 20,而是得到了化合物 19.作者受 Trost 合成(−)-perophoramidine 工作的启发 [2],在吲哚环 4 位引入了溴原子,把 3 位的甲基改为 TBS 取代的乙氧基,用 carreira 配体 L1 进行反应,取得了良好的效果。
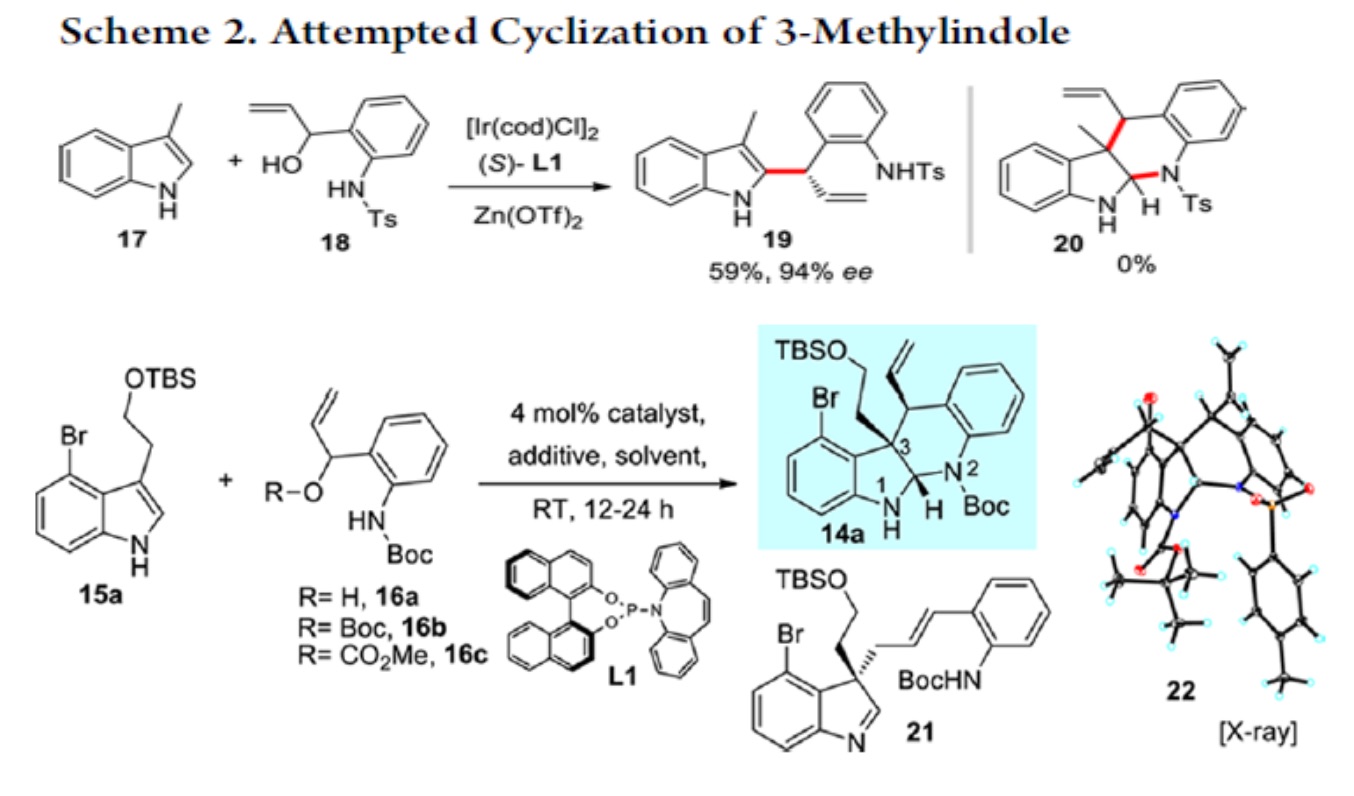
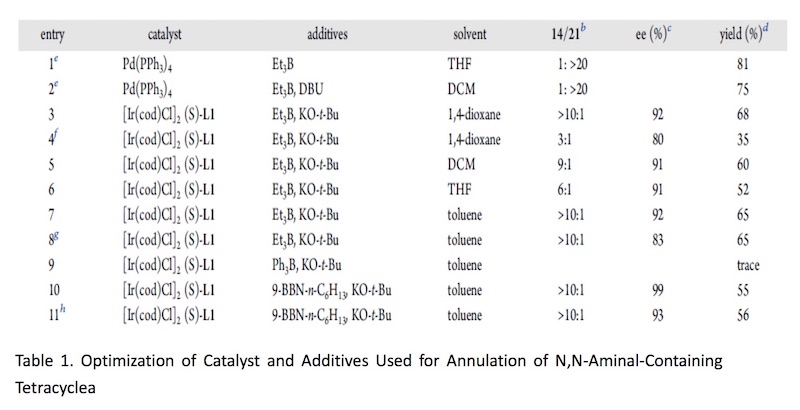
根据 Table1 可知,Pd 催化剂催化时主要以 21 为主,铱催化剂催化时才能实现主要 以 14a 为主。如果按经典的 Tsuji-Trost Reaction 机理,吲哚环应以 3 位进行反应,因为这才 是一个好的 nucleophile。事实上,21 的生成正反映这个事实。当使用铱催化剂时,则不同 于这种情况,如 s’cheme2 。即使并未得到目标化合物 20,但是这一事实却反映了铱催化下 反应不同于 Tsuji-Trost Reaction,或者说不满足于经典的 Tsuji-Trost Reaction 机理。我此处讨 论的目的实际上是想说:scheme 1 所反映的机理是符合经典的 Tsuji-Trost Reaction 机理的, 文中这一过程是有别于经典的 Tsuji-Trost Reaction 机理的。用这个图描述这一过程是否合适? 我们如何考虑这一过程呢?
将 16N 上的-COOMe 为 Boc 后,undesired 产物 21 增多,说明 N 上取代基的空阻对反应的区域选择性有较大影响。得到目标化合物 14a 后,开始全合成。
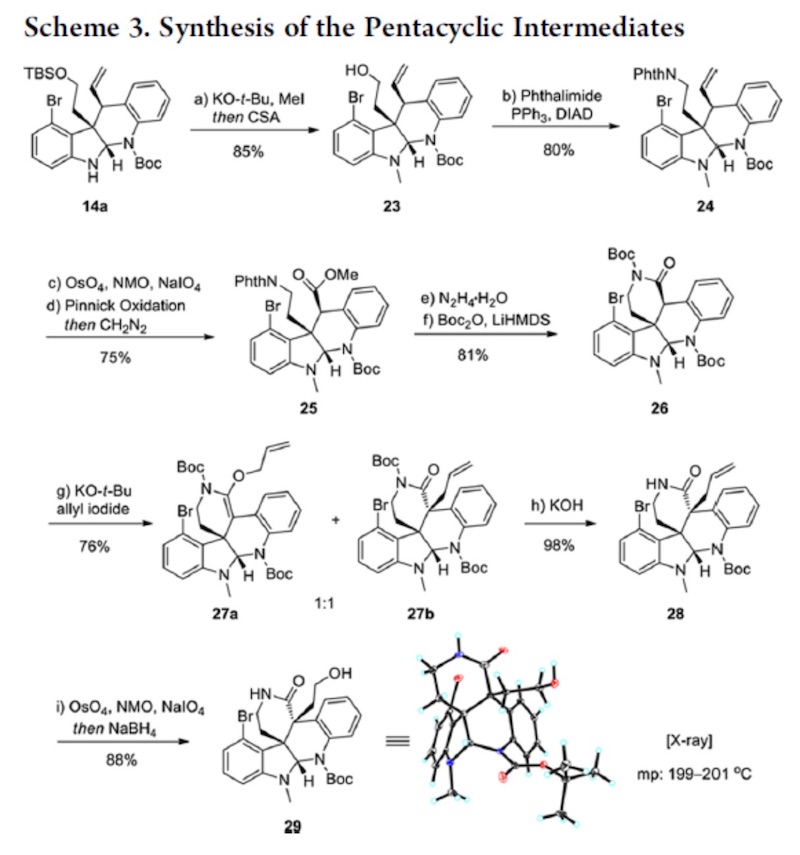
首先,使用使用叔丁醇钾拔去吲哚环 N 上的 H,用碘甲烷进行甲基化。再用 CSA 脱去 TBS 保护基。暴露出醇羟基后采用 Mitsunobu 反应将羟基转化为了 N 取代基。随后,利用 Lemieux−Johnson oxidation 把双键先转化为醛又将醛进行 Pinnick oxidation 。这是一个将双 键转化为羧酸的好方法,在很多合成中都进行了使用。产生的羧酸则用重叠甲烷进行烷基化。 下一步则为后续反应埋下了伏笔。经典的 Gabriel synthesis 使用的是强碱,这并不是一个好 的条件,因为很多底物再此条件下不兼容。所以这里采用的是 Ing-Manske procedure。Phth 掉下后发生的是一个自发的内酰胺化反应。这里考虑为什么会自发?酰胺化后再次用 Boc 保护。对内酰胺进行烷基化,得到的是两个异构体,但无需分离,用叔丁醇钾脱掉 Boc 后, 一加热,O-烷基化产物很容易就[3+3]转化为 C 烷基化产物。这里有两个问题。一是为何连 续的两步先上 Boc,让后再把它脱下来?二是[3+3]为何容易发生?第一个问题,Boc 是否是
没必要上去?其实是很有必要的。因为 Phth 掉下之后,内酰胺化是一个平衡反应,一个平 衡反应是需要有推动力才能发挥其作用,而这里缺少的就是推动力。Boc 是推动力吗?可以 这样理解,但不准确。形成酰胺后,酰胺很容易经过一个平衡过程又回去了,而 Boc 上去之 后,酰胺在反应回去的过程就被打破了,其结果就是推动了反应的进行。第二个问题 Boc 下来之后实际上是相当使酰胺的活性又解放出来,同时在强碱下,N 上的 H 又会被拔出,是 [3+3]结构富电子话,这实际相当于活化了[3+3]反应体系。这一理念其实在几个 claisen 反应 中体现的淋漓尽致,如 Eschenmoser-claisen,Ireland-claisen,Johnosen-claisen。内酰胺形成 后再一次 Lemieux−Johnson oxidation。随后 Mitsunobu,Heck,SN’2, 得到 33 后,离 Communesin F 仅有一步,然而经过尝试,33 难以转化为 Communesin F。为了避免经过 33, 作者改变策略,经 35 合成 Communesin F。
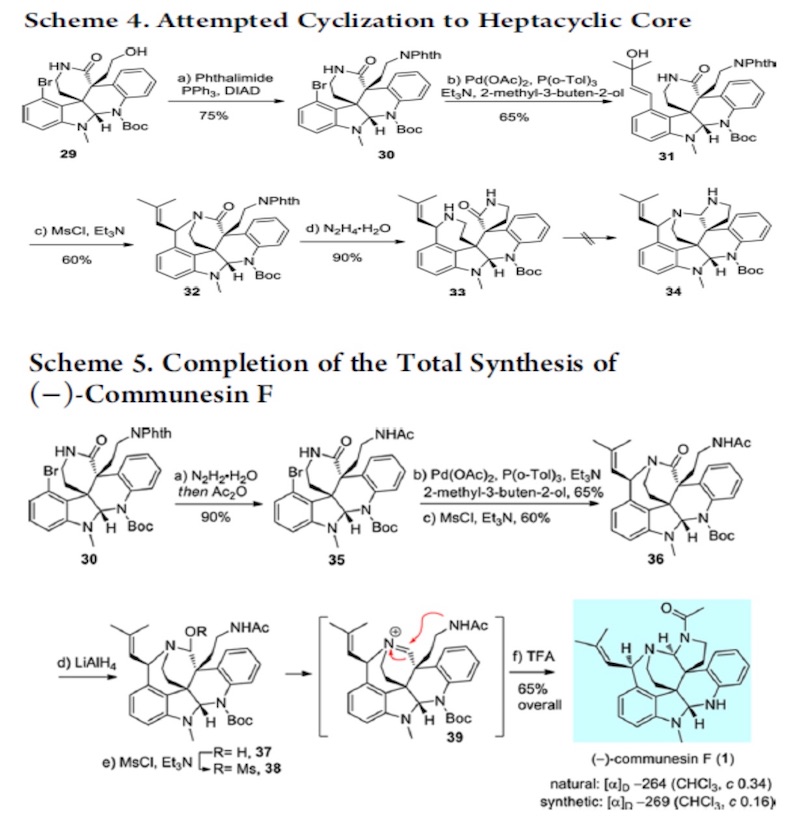
这里要讨论的是为何不能经过 33 而要用 Scheme 5 才能合成?文中并未做任何解释,这 里详细讨论。之前讨论过一个问题自发的内酰胺化反应为什么会自发?就是因为形成后可以 减小产物空阻。形成 33 后导致其不能形成 Communesin F 的一大原因也是空阻,但不能理解为简单的空阻,我想,这也是不同水平之间最清晰的区别,虽然都说空阻,但理解的深度不 同。最简单的空阻原因就是由于羰基周围空阻大影响了亲核试剂对羰基的进攻,这是一部分 原因,另一个空阻指的是是羰基为 SP2 杂化,平面构型,空阻较小,反应后则形成 SP3 杂化, 四面体构型,空阻变大。这个原因导致了羰基的惰性。这一现象与李昂老师 clostrubin 合成中遇到问题如出一辙。[3]另一个原因是 N 位于六元环上,自由度减小,这也限制了从 33 合成 Communesin F。Scheme5 中的路线,先构筑 G,B 环,首先这在空阻上就是有利的, 同时缩醛氨还具有形成亚胺正离子的倾向,进一步推动了 反应的进行。其次是 N 取代基位于支链上,自由度大,有 利于反应的进行。下图为上述解释的 3D 图。
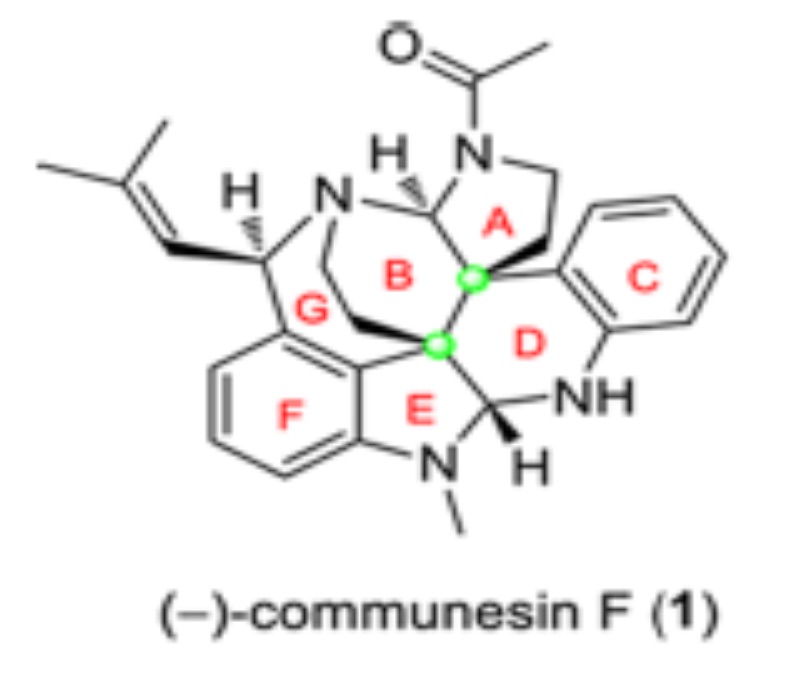
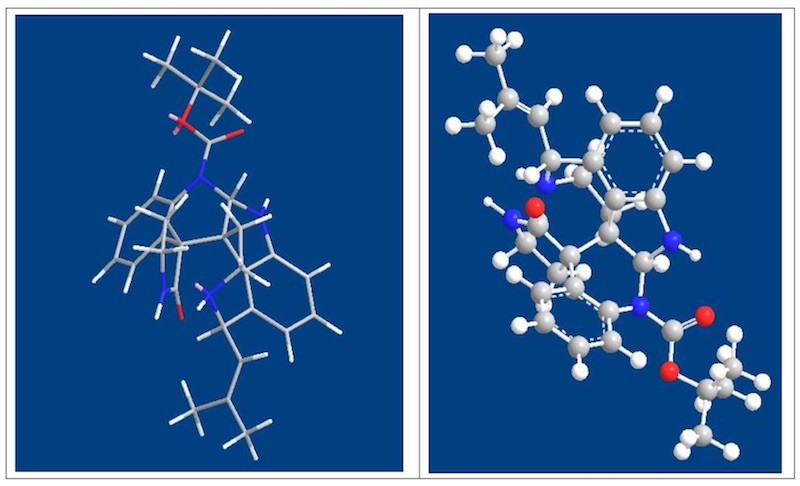
总之,这一合成利用铱催化的不对称合成方法学简洁有力的通过一步反应构筑出一个四 环体系,同时,最后几步巧妙而漂亮的完成了 Communesin F 合成。
参考文献及链接
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!















No comments yet.