
本文投稿作者 alberto-caeiro
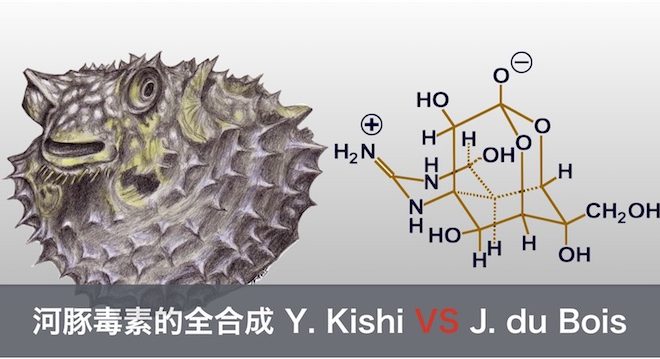
河豚鱼作为一种日本特别的烹饪食材而闻名。但如果处理不当,一餐的河豚鱼足以使餐桌上的各位食客丧命。于是在日本,能够烹饪河豚鱼的大厨都需要学习考试,以获得特殊的许可证。
19世纪开始,日本就有许多生物学家开始研究这种剧毒的物质。1911年,Tahara报道了这种剧毒的提纯物,并将其命名为tetrodotoxin[1]。虽然在后来证明其中只含有少量的tetrodotoxin,但其关键性的分离为日后的工作提供了许多帮助,并在当时的药学研究提供了基础。在1952年,津田和川村也分离出这种化合物,并且沿用了tetrodotoxin [2]。在第三届IUPAC(Kyoto Japan)上,Tsuda[3],Goto[4],Woodward[5]都展示了各自得到河豚毒素的结构式,三者同时且独立的得到了河豚毒素的结构式,如下图1所示。

图1:tetrodotoxin三种形式
河豚毒素是一种神经毒素,对钠离子通道有抑制作用。河豚毒素能够抑制钠离子通道的开放,故可以高效的关闭周围神经系统。根据食用量的不同,症状从麻痹、昏迷、呼吸衰弱、死亡依次递增。有趣的是,河豚毒素对大脑神经没有直接作用,中毒者直至死亡时才有意识到中毒。其选择性的作用机制使得其在药学和生物化学上有许多研究。
Kishi的全合成研究[6,7]
1)逆合成分析
得益于在Goto组关于河豚毒素结构的研究,Y. Kishi得到许多河豚毒素物理和化学性质的第一手资料,这些都被有效的运用到其合成策略中。下图2为Y. Kishi组的逆合成分析。
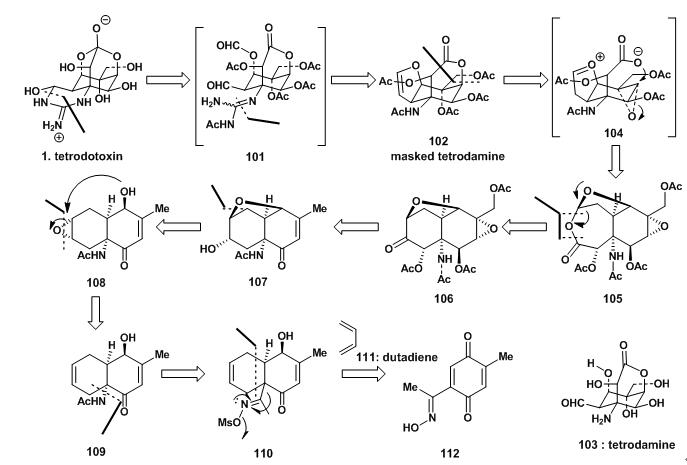
图2:Kishi’s retrosynthesis of Tetrodotoxin
因河豚毒素易溶于水而难溶于有机溶剂,于是构想切掉胍基后的物质再经乙酰基修饰后得到102这种方案,而在Goto组已得到证明,虽然河豚毒素碱性条件下不稳定,但乙酰基保护的衍生物可以在温和条件下可以去保护。
2)Tetrodotoxin的全合成
底物113与丁二烯直接发生D-A反应时,肟基是给电子基团,反应只会得到热力学控制的产物114。于是,Y. Kishi通过加入Lewis酸二氯化锡,得到同面反应的产物116。其中可能经过115这种中间产物,金属锡与肟基和羰基形成六元环中间产物,此时的肟基变成给电子基团,从而得到需要的116。
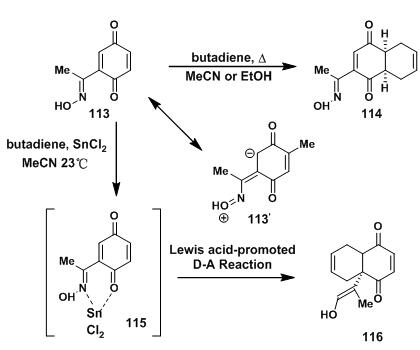
图3:Kishi’s D-A model studies
117先与羟胺形成肟后经氧化银氧化形成苯醌衍生物112;112经lewis-acid promoted D-A反应得到119;110经Beckmann rearrangement得到121,并且构型得到保持;121经选择性还原羰基得到109;109中双键得环氧后同时发生分子内的另一个环氧开环与环氧化的串联反应得到117。

图4:Kishi’s synthesis of cis-decaliin system 107
107中游离的羟基经氧化成羰基后用乙二醇保护得122;122中另一个羰基经Meerwein-Ponndorf-Verley Reduction后,用乙酰基保护得单一构型的123;123中双键上的甲基经二氧化硒氧化得α, β-不饱和醛[8](a review on selenium dioxide oxidation),后经还原得到烯丙基醇124;124中双键环氧化得126,但在其中,此双键非常难以反应,需要升高温度,而升高温度会导致m-CPBA的分解,为此Kishi筛选了许多自由基抑制剂,最终选择了125,并且得到不错的收率;126中羰基换成用乙醇保护得128;二乙氧基缩酮128在180℃下分解得129。
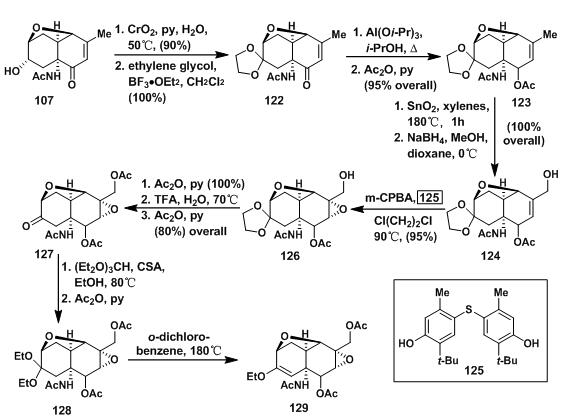
图5:Kishi’s synthesis of enol ether 129
129经环氧化得130;130经乙酸催化得串联反应得到106;106经Baeyer-Villiger oxidation 得105;105在酸性条件下先形成氧鎓离子,打开7元环内酯,后与另一个环氧化合物发生环氧开环反应得到另外一个环内酯135;135中羟基用乙酰基保护后,加热脱去一分子乙酸得到乙酰基保护的tetrodamine 102。至此,tetrodotoxin的基本环骨架已得到构建。
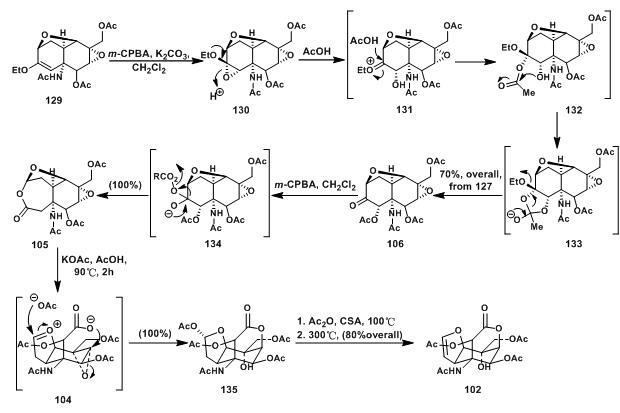
图6:Kishi’s synthesis of masked tetrodamine
102在烷基化试剂Meerwein’s salt 作用下,生成136,后在酸性条件下水解得137;137与138反应生成139后继续与乙酰胺反应生成胍基化合物140;140与氨气发生胺交换生成141;141经四氧化锇氧化生成邻二羟基化合物142,后经高碘酸氧化得101,弱碱性条件下发生亲和反应成最后一个含氮环并且水解去乙酰基保护,以25%的4步收率,得到最终的tetrodotoxin。

图7:Kishi‘s total synthesis of (+) tetrodotoxin
J. du Bois的全合成研究[9]
1) 逆合成分析
在进行tetrodotoxin的全合成研究之前,Du Bois小组已经发展了一套Rh氮卡宾(Rh nitrenoid)插入的方法学[10](a review on carbene and nitrene C-H insertion reaction),于是在此工作中多次用到此方法对C-H键进行官能团化。
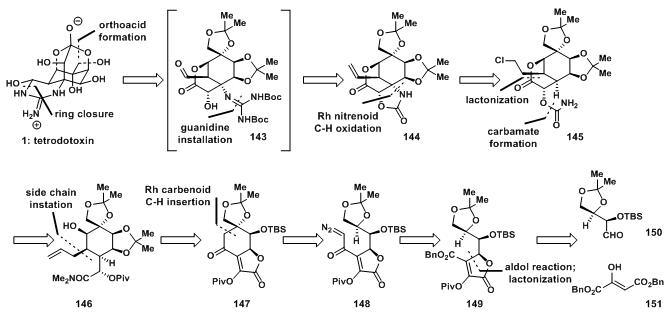
图8:Du Bois’s retrosynthetic analysis
2)Tetrodotoxin的全合成
152先环氧化后发生亲和的串联反应,后经皂化得到156;156先开内酯环得酰胺后,再用二甲氧基缩丙酮保护邻位二醇得157,157再用TBSCl保护二级羰基得158;在158还原时,容易发生160所示得断键方式,得到三级胺,最终Du Bois采用加入n-BuLi形成159所示结构,后成功得到醛150;

图9:Du Bois’s synthesis of 150
150与151发生aldol反应得到中间产物162,形成得氧负离子立刻发生环内酯化得到环内酯163,其中,得益于Felkin-Ahn control[11]反应条件,163的dr值为12:1;164经过钯碳氢还原后,8a位上的酯基变成羧基后,在草酰氯的催化下与重氮甲烷发生亲核取代反应得到148;再用Du Bois组发展的方法学,进行特定的卡宾C-H键插入反应,得到147;147再还原得到165。
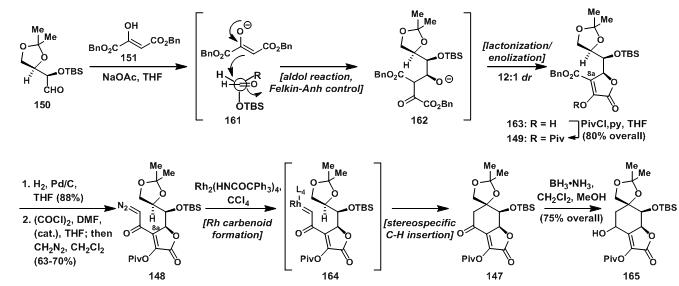
图10:Du Bois’s synthesis of 165 through a C-H insertion
虽然4取代双键很难被还原,但在铑碳氢、高压和TFA的作用下,依然可以得到同面加成的产物166,166立刻发生酯交换反应,得到张力更小的167,再重新上保护基得168;168先开环生成羟基后被NMO氧化得到羰基[12],之后发生Takai methylenation得到170[13];后在171得催化下,氧化得到α, β-不饱和酮[14];172与乙烯基格氏试剂反后,还原得到146;146接着发生分子内酯化反应,后脱除羟基保护得174。
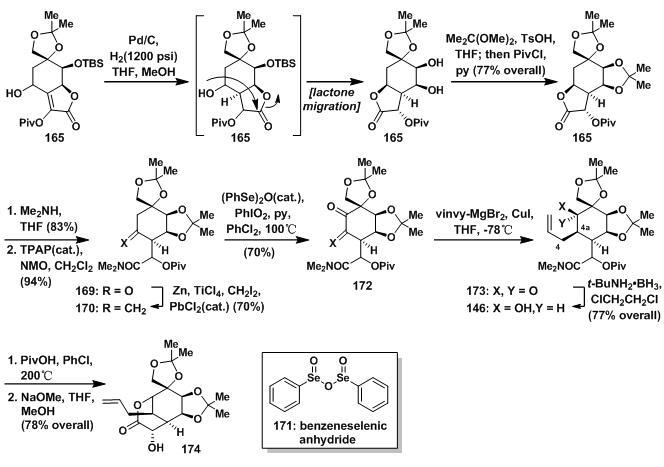
图11:Du Bois’s synthesis of lactone 174
174中自由的羟基先与异氰酸酯175反应生成酯酰胺,再在锌的作用下消除三氯乙酰基得176;176先臭氧化后还原得到得羟基再与MsCl,吡啶反应得145;145发生另一次铑卡宾C-H键插入得179;179先发生SN2生成硒醚,再发生氧化消除得到144;180发生水解反应即可得到181;184先臭氧化得到醛基后,在TFA的作用下可以得到最终产物,但是产物为1和2的混合物,比例为1:1。
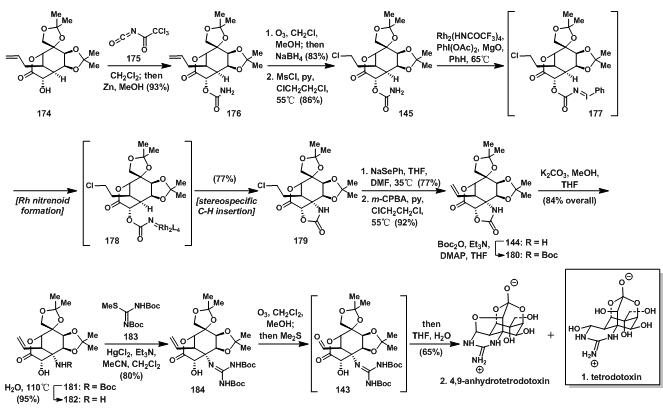
图11:Du Bois’s total synthesis of tetrodotoxin
写在最后的话
1972年,Kishi对tetrodotoxin的合成是一项卓越的工作,他们对立体化学的精准控制以及对化学反应的准确应用,令人叹为观止。另一方面,第一手文献知识的重要性在这里也有体现。2003年Du Bois通过C-H键官能团化重新完成了此全合成工作,到现在,C-H键官能团话在全合成中占据着重要的作用。
参考文献
- Tahara, et. al. Biochem. Z., 1911, 30, 255;
- Tsuda, et. al. J. Pharm. Soc. Jpn., 1952, 72, 711;
- Tsuda, et. al. Chem. Pharm. Bull., 1964, 12, 1357;
- Goto, et. al. Tetrahedron, 1965, 21, 2059;
- B. Woodward, Pure Appl. Chem., 1964, 9, 49;
- Kishi, et. al. J. Am. Chem. Soc., 1972, 94, 9217;
- Kishi, et. al. J. Am. Chem. Soc., 1972, 94, 9219;
- Rabjohn, Org. React., 1976, 24, 261;
- Du Bois, et. al. J. Am. Chem. Soc., 2003, 125, 11510;
- a) M. A. Mckervey, al. Chem. Rev., 1994, 94, 1091;
- b) R. E. J. Beckwith, al. Chem. Rev., 2003, 103, 2861;
- Reiser, et. al. Chem. Rev., 1999, 99, 1191;
- P. Marsden, et. al. synthesis, 1994, 639;
- Takai, et. al. Tetrahedron Lett., 1978, 19, 2417;
- Crich, et. al. Tetrahedron, 1985, 41, 4359;
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!














No comments yet.