
本文作者:杉杉
导读
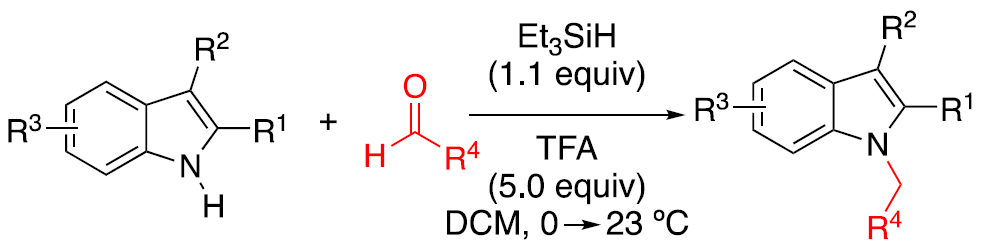
本文主要报道了在无金属催化剂的条件下,以醛为烷基化剂,Et3SiH为还原剂,成功实现了吲哚的还原性N-烷基化反应。同时,该反应具有良好的底物范围,各种取代的芳香族/脂肪族醛和吲哚均为合适的底物。此外,该反应可进行一锅串联1,3-烷基化反应从而合成双烷基化化合物。其中,德克萨斯大学Doug E. Frantz和 百时美施贵宝Albert J. DelMonte为共同通讯作者。
A Metal-Free Reductive N‑Alkylation of Indoles with Aldehydes
Nicholas A. Clanton, Taylor E. Spiller, Eliezer Ortiz, Zhinong Gao, Juan Manuel Rodriguez-Poirier, Albert J. DelMonte,* and Doug E. Frantz*
Org. Lett. 2021, 23, 3233-3236. DOI: 10.1021/acs.orglett.1c00179.
正文
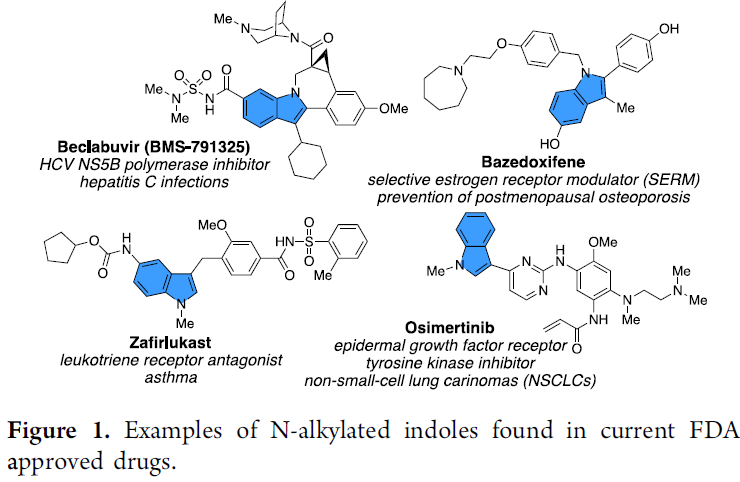
吲哚环骨架广泛存在于各类药物和天然产物中,其中氮取代的吲哚环化合物也备受关注,如美国食品药品监督管理局(FDA)批准的药物中也包含此类骨架(Figure 1)。因此,对于氮取代吲哚环的合成,具有重要的意义。
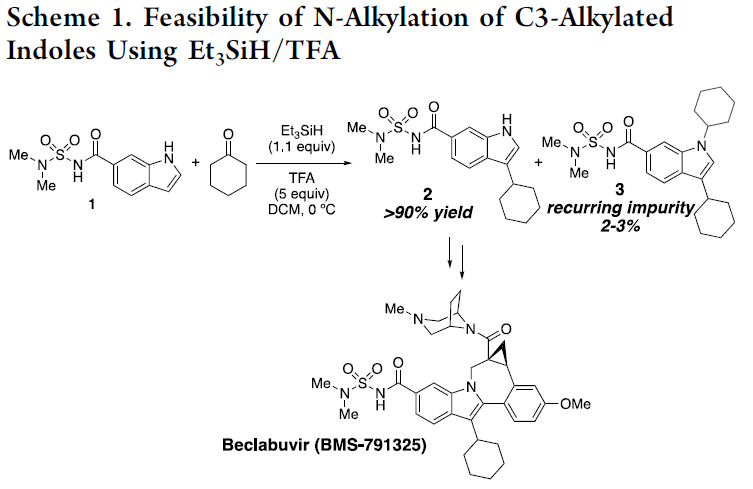
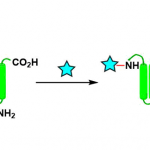
然而,由于吲哚中氮原子的亲核性较弱,对于吲哚环的N-烷基化反应,具有一定的难度。为了增加亲核性,已使用强碱进行去质子化从而实现活化,但也存在一定的局限性,如与碱敏感性底物不兼容、依赖有毒的烷基化试剂等。此外,也可通过Mitsunobu型方法将醇用作烷基化试剂。同时,醇也被进一步用于“借氢”策略中,通过反应性更高的二氢吲哚中间体实现吲哚的N-烷基化反应。如Seidel等[1] 开发了一种吲哚N-烷基化的方法,涉及衍生自二氢吲哚和醛的瞬态叶立德的氧化还原异构化过程。对于传统的有机金属方法也备注关注,如Hartwig[2]和Trost[3]等报道关于铜催化吲哚和N-甲苯磺酰的偶联反应或π-烯丙基化学反应。最近,通过使用手性过渡金属催化剂也实现了吲哚的不对称N-烷基化反应[4-6]。此外,由于APIs中对于残留金属的量需严格控制,若能通过一种无金属催化的策略,则更具吸引力。受Appleton等[7-9]工作的启发,作者以环己酮为底物,成功在吲哚的C3-位进行烷基化,同时也获得少量的双烷基化副产物3(Scheme 1)。因此,对于C3-烷基化的吲哚底物,可在还原性胺化条件(Et3SiH/TFA),进一步与羰基衍生物进行N-烷基化反应。
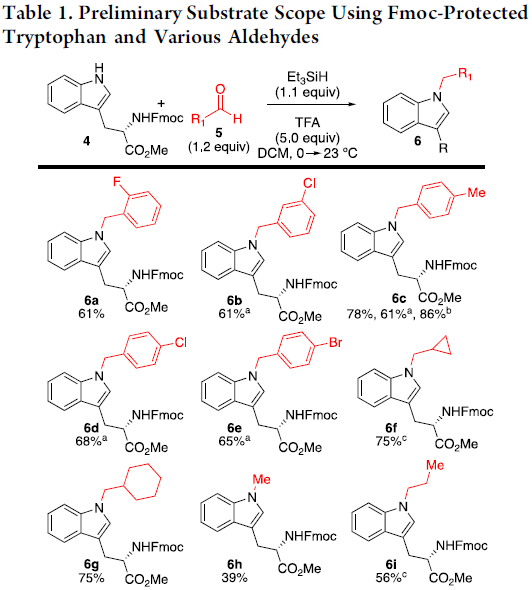
首先,作者选择C3-烷基化的吲哚4作为底物,对醛的范围进行了扩展(Table 1)。具有不同取代的芳基醛,均可顺利反应,获得相应的产物6a–6e。同时,一系列脂肪醛也与体系兼容,获得产物6f–6l。值得注意的是,多聚甲醛也可作为甲基化剂,获得产物N-甲基化产物6h。对于6c的克级实验,同样取得预期的结果,收率为86%。
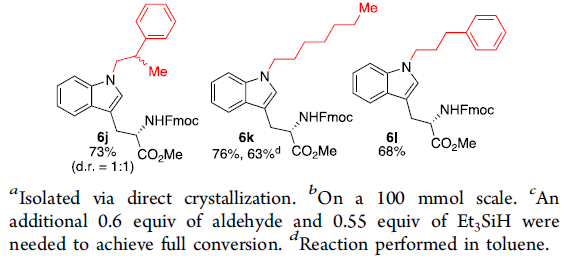
随后,作者选择了几种不同C3-烷基化的吲哚为底物(Table 2),如咔唑均可顺利反应,获得产物8a–8e。同时,吲哚的C3-位上具有羰基官能团时(如酮和酰胺),也与体系兼容,获得产物8g和8h。此外,吲哚的C3-位上为环己基时,可获得产物8f和8i。
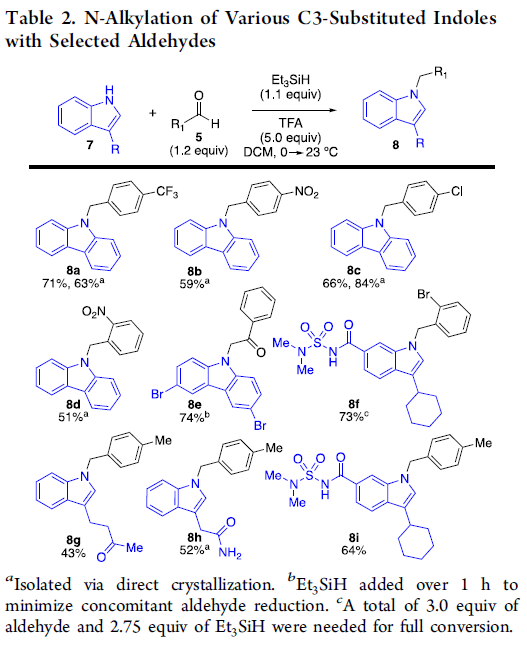
此外,作者对反应的实用性进行了相关的研究(Scheme 2)。首先,醛5a可与吲哚4进行N-烷基化反应,合成化合物9,作为亲电试剂用于结构-活性关系(SAR)的研究。其次,醛5b与伯叠氮化物可通过点击化学作为生物连接物,用于生物活性吲哚的细胞靶标鉴定研究,可合成化合物10和11。此外,药物佐米曲普坦(zolmitriptan)也可直接进行N-烷基化反应,获得化合物12。同时,以beclabuvir(1)与环己酮和对甲基苯甲醛为底物,可通过一锅串联C3-烷基/N-烷基化反应,从而以55%的收率得到双烷基化吲哚8i。
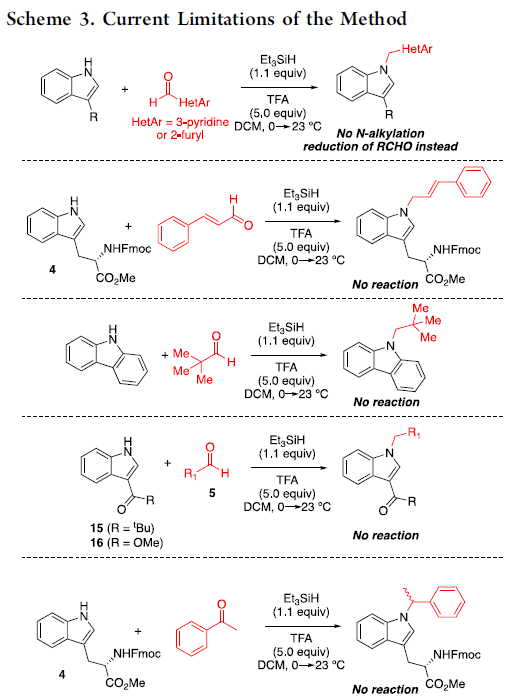
随后,作者还对反应的局限性进行了研究(Scheme 3)。当以杂芳基醛为底物(如3-吡啶甲醛和2-糠醛),未能获得N-烷基化产物,可能是由于醛被还原成醇并与N-烷基化反应存在动力学竞争关系。其次,α,β-不饱和醛(如肉桂醛)也不能顺利反应。对于具有空间位阻的醛(如戊醛),也未能进行反应。此外,当吲哚的C3-取代为酰基取代(如15和16)时,由于亲核性降低,导致反应也未进行。对于酮底物(如苯乙酮)也未能和吲哚底物4顺利反应。
总结
本文主要报道了在无金属催化剂的条件下,以醛为烷基化剂,Et3SiH为还原剂,成功实现了吲哚的还原性N-烷基化反应。同时,该反应具有广泛的底物范围、良好的官能团耐受性、温和的反应条件等特点。此外,作者还对反应的局限性也进行了相关的研究。














No comments yet.