

译自Chem-Station网站日本版 原文链接:ゼロから始める!量子化学計算~遷移状態を求める~
翻译:炸鸡
今天以丙二醛分子内的氢迁移反应为例,介绍怎么用Gaussian和GaussView求解反应的中间体能量值。
适读人群
・有Gaussian软件但还不知道用法的人
・想入门量子计算化学的人
计算流程
求解的氢迁移反应如下。
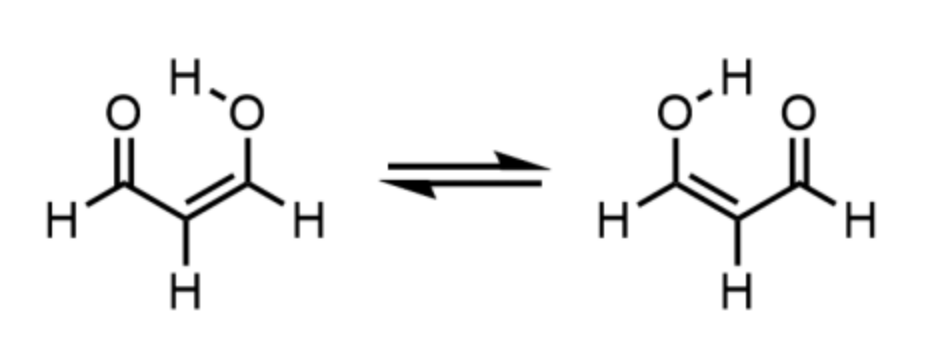
图 1. 氢迁移反应
首先,在GaussView上画出丙二醛的结构。从苯环开始画再删掉一些多余的原子会比较省力。
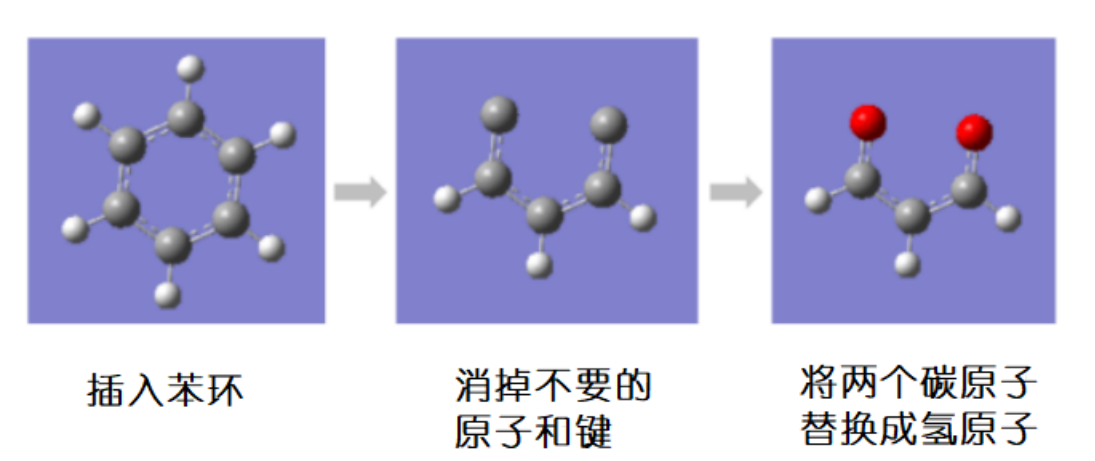
图 2
把要转移的氢原子挪到设想的中间体中的位置。放的位置越接近真实中间体里转移氢的位置计算结果越准确。首先在环的内侧放置一个氢原子,之后加上氢氧键。
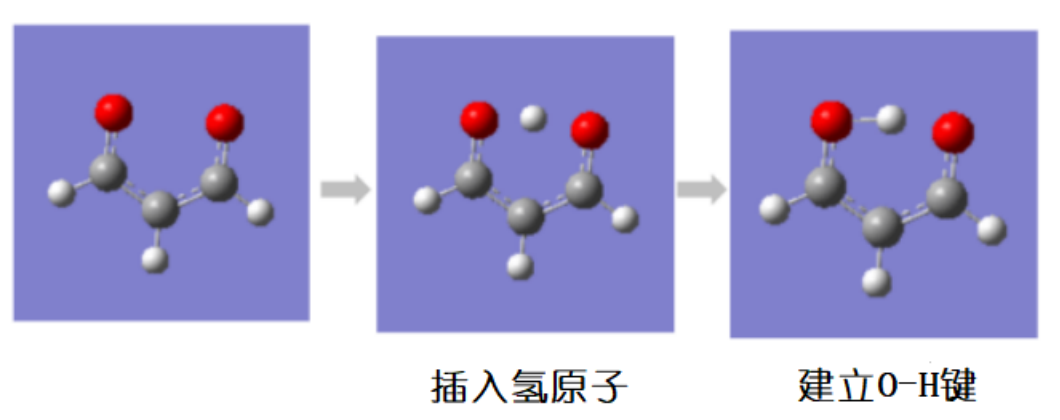
图 3
接下来建立inputfile。Job Type设定为Optimization+Frequency。Optimization表示结构优化,Frequency表示振动分析。“Optimize to a”选择TS(Berny),向软件发出求解中间体的指示。“Calculate Force Constants”选择Once,计算方法选择Hartree-Fock。以上设置完毕,开始计算。
先打开log文件内result的vibrations,确认振动数(Freq)为负值,只有为负值,所画的分子有局部稳定的结构,才可能有中间体。选择振动数为负,之后进入下一步。
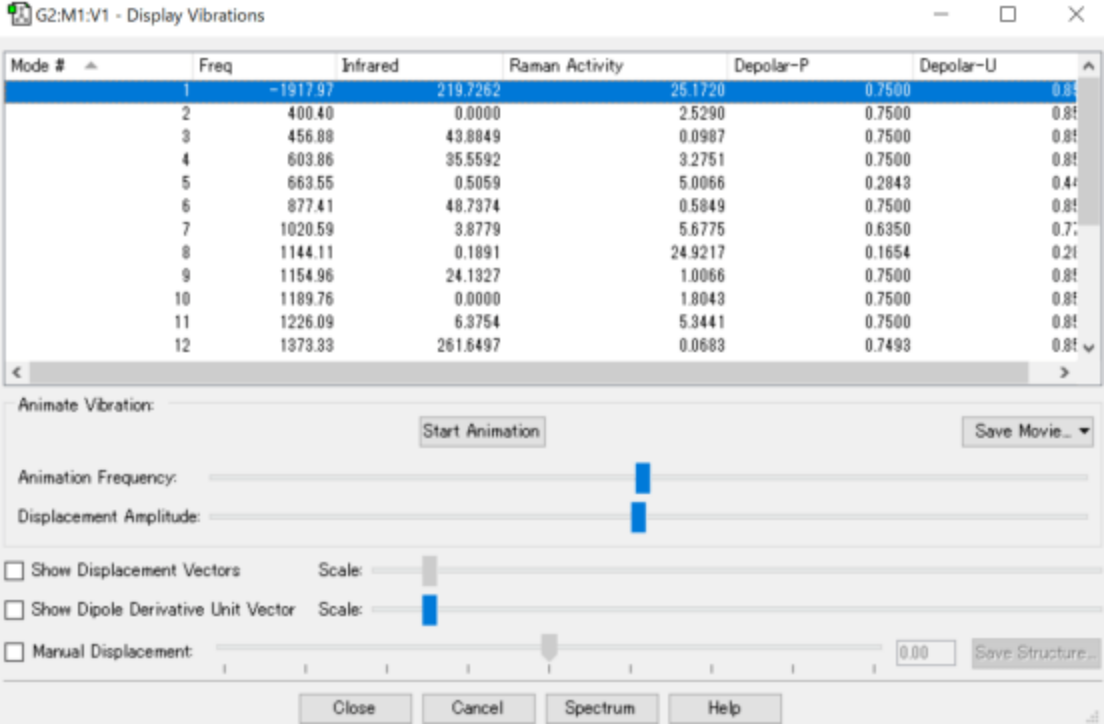
图4
Job Type中选择IRC。这是用来确认中间体是不是反应物到生成物中间的状态(本次计算指氢原子移动前和移动后)。选择Compute more points可以改变计算范围。默认设定为10。这次初始设定为30。Guess和Solvation设为默认,Additional Keywords这里暂不需要填写。计算结果如下图所示。
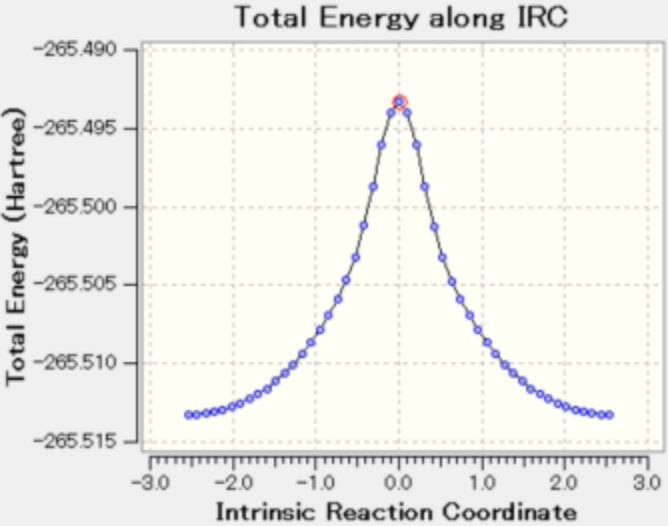
图 5
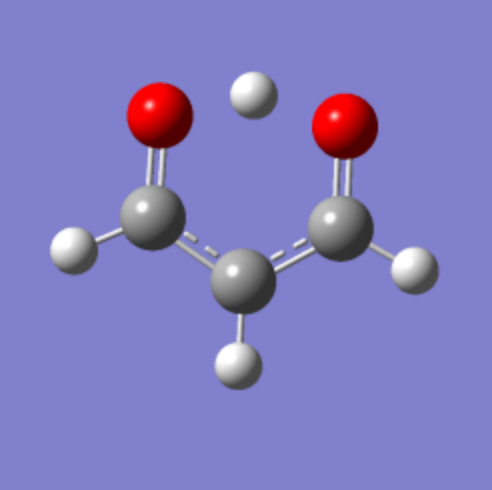
图 6
图5 展示了整个分子的能量变化,能量越高结构越不稳定,能量越低结构越稳定。中间体时能量最高。中间体时的丙二醛分子的转移氢位于两个氧原子的正中间。
图7是图5的微分形式(微分能量的平均值),纵轴的0表示能量的最高值。
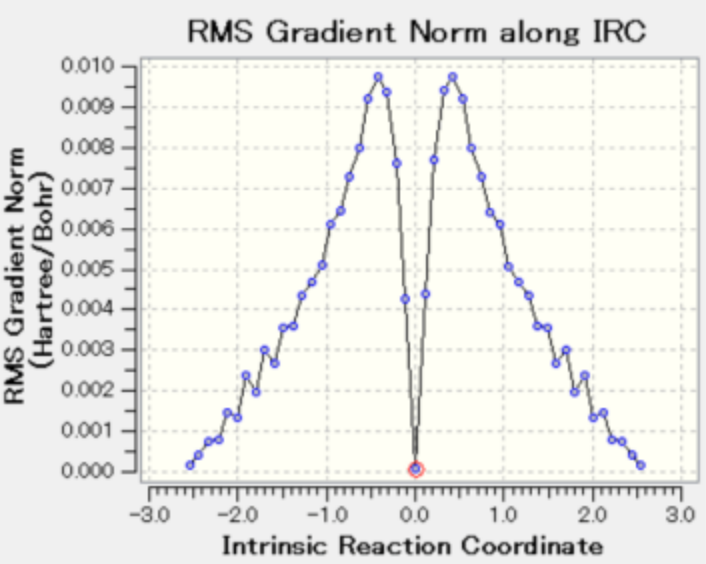
图 7
应用
联想以下丙二醛分子内氢转移的反应过程,不难理解能量变化图的曲线是对称的。如果将丙二醛碳链上的一个氢原子替换成其他原子,会发生什么有趣的现象呢?
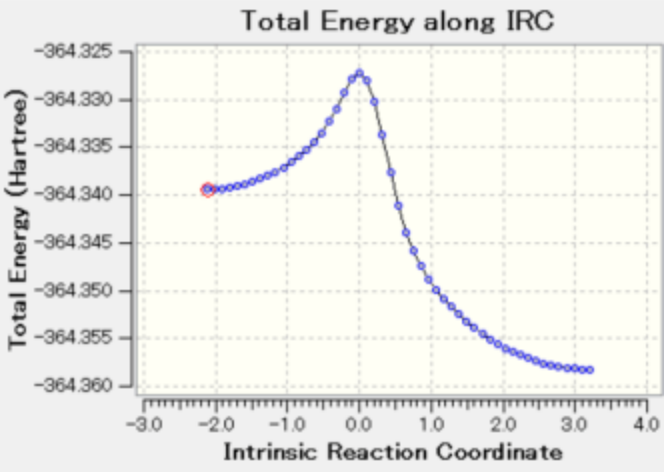
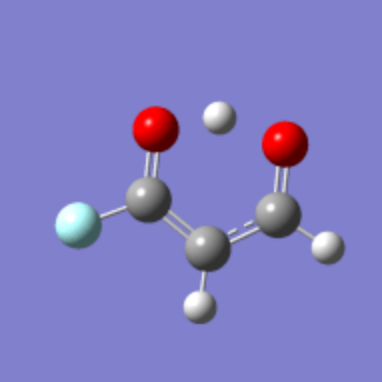
图 8. 将一个氢原子替换成氟原子后
如图 如果将其中一个醛基的氢换成氟,曲线图就不对称了。
后记
我用Gaussian的时候经常需要请教老师。量子化学不同于常用的电子计算器,计算过程可谓是充满了弯弯绕绕。本篇记事虽然谈不上什么深入挖掘﹑手把手教学,只是一个备忘录性质的记事,以供那些想学习用软件进行量子化学计算但不知道怎么开始动手的人一点微薄的帮助。希望你们喜欢。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载


















No comments yet.