
作者:石油醚
导读:
南开大学化学学院朱守非课题组发展了一例铁催化1,3-烯炔的烯丙位C(sp3)‒H键硅化新反应。机理研究发现,该反应经历一种新型C‒H键活化模式,自旋交叉在该反应中可能具有多重调控作用,即自旋交叉效率决定了反应的区域选择性、自旋交叉可在催化历程的不同阶段阻碍或加速反应。相关成果发表于Angew. Chem. Int. Ed.上(DOI: 10.1002/anie.202402044),文章第一作者是南开大学博士研究生何鹏。
“Iron-Catalyzed Allylic C(sp3)‒H Silylation: Spin-Crossover-Efficiency-Determined Chemoselectivity
Peng He (何鹏), Mu-Han Guan (管沐涵), Meng-Yang Hu (胡梦阳), Yuan-Jun Zhou (周塬峻), Ming-Yao Huang (黄明耀), Shou-Fei Zhu (朱守非)*
Angew. Chem. Int. Ed. 2024, e202402044. DOI: 10.1002/anie.202402044”
正文:
从自旋的角度出发,过渡金属催化剂可分为闭壳层催化剂和开壳层催化剂,以铁等3d金属配合物为代表的开壳层催化剂电子自旋态丰富,反应途径多样,常常表现出异于传统闭壳层催化剂的性质,具有重要发现机遇。揭示开壳层催化剂的自旋效应是提升地壳丰产金属催化剂理性设计水平的关键,还有望突破闭壳层催化剂的局限,为催化领域带来颠覆性变革,具有重要研究价值。然而迄今人们对自旋效应的理解非常有限,阻碍了开壳层催化剂的理性设计。自旋交叉是一种重要的自旋现象,然而目前人们对自旋交叉影响化学反应的认识主要局限于其可降低反应能垒(图1A),对其更多维度的认识将为理解和应用开壳金属层催化剂的自旋效应带来重要突破。
南开大学朱守非教授课题组长期关注开壳层催化研究,该课题组近年来发展了一系列新型开壳层铁系金属配合物催化剂,并将其成功应用于多种不饱和烃的氢/碳金属化反应以及交叉偶联反应,展现出明显的不可替代性(J. Am. Chem. Soc. 2024, 146, 5051;
Nat. Sci. Rev. 2024, doi: 10.1093/nsr/nwad324; Angew. Chem. Int. Ed. 2023, e202315473; Chin. J. Chem. 2023, 41, 3547; J. Am. Chem. Soc. 2022, 144, 515; ACS Catal. 2022, 12, 2581; Chem. Sci. 2022, 13, 2721; Chem. Sci. 2022, 13, 7873; J. Am. Chem. Soc. 2020, 142, 16894; J. Am. Chem. Soc. 2019, 141, 4579; Nat. Commun. 2018, 9, 221)。其中,在邻菲罗啉/铁配合物催化的炔烃区域发散性硅氢化反应中(J. Am. Chem. Soc. 2020, 142, 16894),他们揭示了催化剂可以通过自旋交叉以及金属与配体间的自旋离域动态调节金属中心的自旋态和氧化态,从而加速氧化加成与还原消除这两个电性需求相反的基元反应(Natl. Sci. Rev. 2024, 11, nwad324)。近期,该课题组又利用上述催化体系发展了一例铁催化1,3-烯炔的烯丙位C(sp3)‒H键硅化新反应,发现了该反应经历一种新型C‒H键活化模式,揭示了自旋交叉在该反应中可能具有多重调控作用,即自旋交叉效率决定了反应的区域选择性、自旋交叉可在催化历程的不同阶段阻碍或加速反应(图1B)。相关成果发表于Angew. Chem. Int. Ed.上(DOI: 10.1002/anie.202402044)。
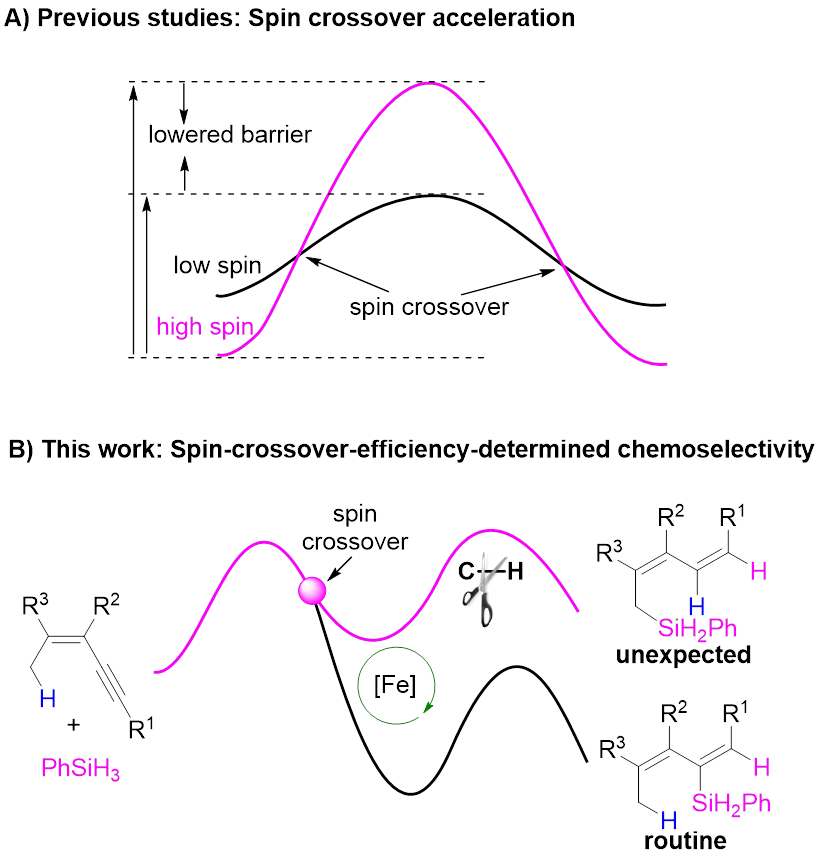
图1 开壳层催化剂的自旋效应
可能由于烯烃相较于C(sp3)‒H键更容易发生硅氢化反应,烯丙位C(sp3)‒H键的直接催化硅基化反应一直没有成功的先例。本文发展的催化体系可实现多种链状和环状1,3-烯炔的烯丙位C(sp3)‒H硅化反应,生成一系列具有共轭二烯结构的烯丙基硅化合物,且产物中烯基的构型都得到很好的控制(图2)。该反应除了生成烯丙位C(sp3)‒H硅化产物,还伴随生成一定量炔烃硅氢化产物,两种产物比例可由配体和底物调控,生成烯丙位C(sp3)‒H键硅化产物的化学选择性最高可达>98:2。C2位芳基上的取代基影响反应速率但不影响化学选择性。将C2位的取代基换为烷基取代基时,反应也能顺利发生,并且该烷基取代基位阻越大,C‒H键硅化选择性越高,当该取代基为叔丁基时,C‒H键硅化选择性>98:2;C2位无取代基时则只能生成炔烃硅氢化产物。炔基末端取代基对反应影响较小。底物中含有环内双键时,环的大小显著影响反应化学选择性,七元环的C‒H键硅化选择性>98:2。环外双键类型底物和苯丙炔类底物则以专一的化学和区域选择性发生炔烃硅氢化反应。
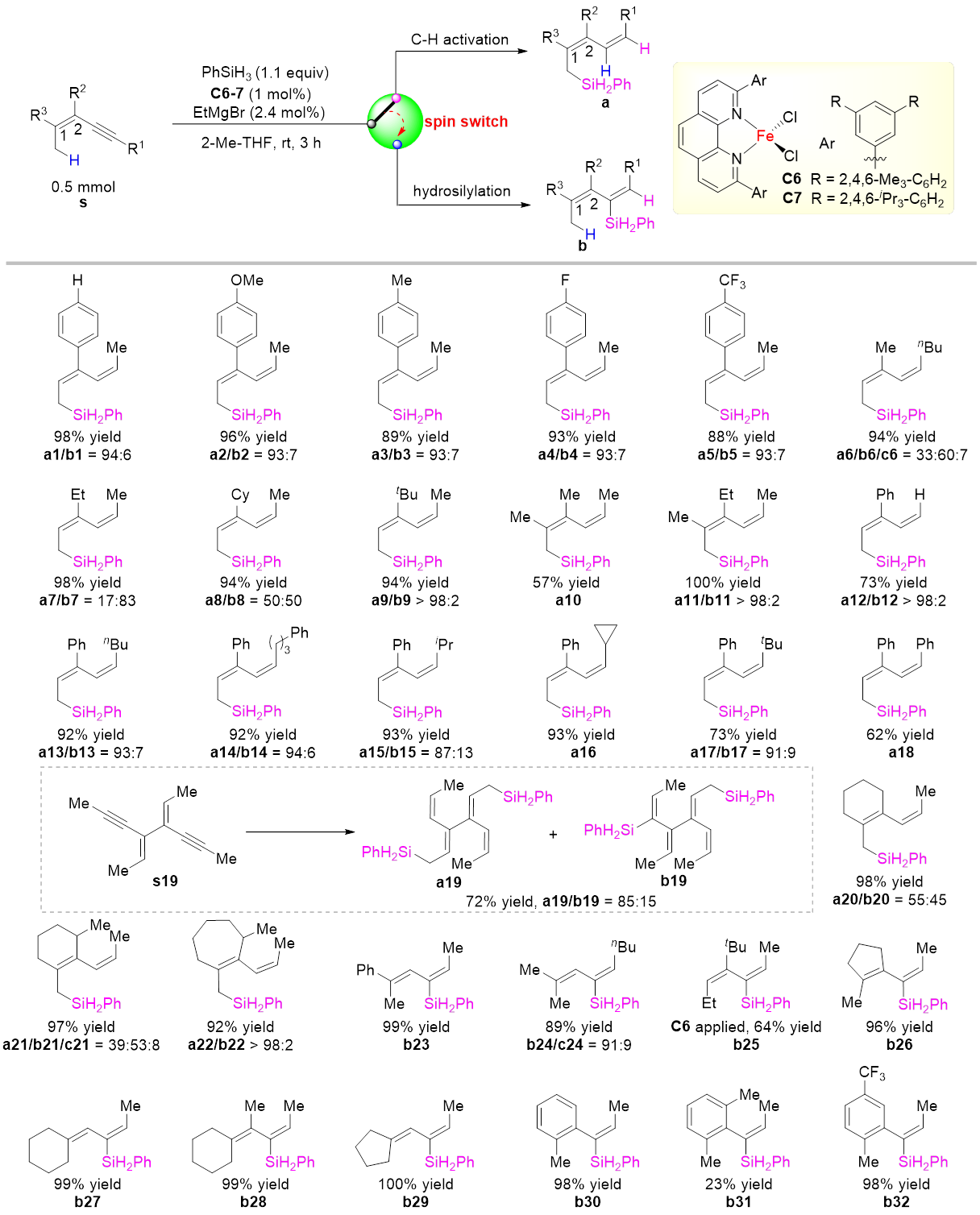
图2 铁催化共轭烯炔的烯丙位C(sp3)‒H硅化和硅氢化反应
同位素标记实验表明,加成到C≡C键上的两个氢原子分别来自Si‒H和烯丙位C‒H(图3A)。混合硅烷实验排除了Fe‒H或Fe‒Si物种启动的反应机理,并支持(形式上)零价铁物种启动的两电子氧化还原机理(图3B)。研究者还制备了(形式上)零价铁活性配合物,并验证了其催化效果与原位还原体系一致,进一步支持上述机理(图3C)。
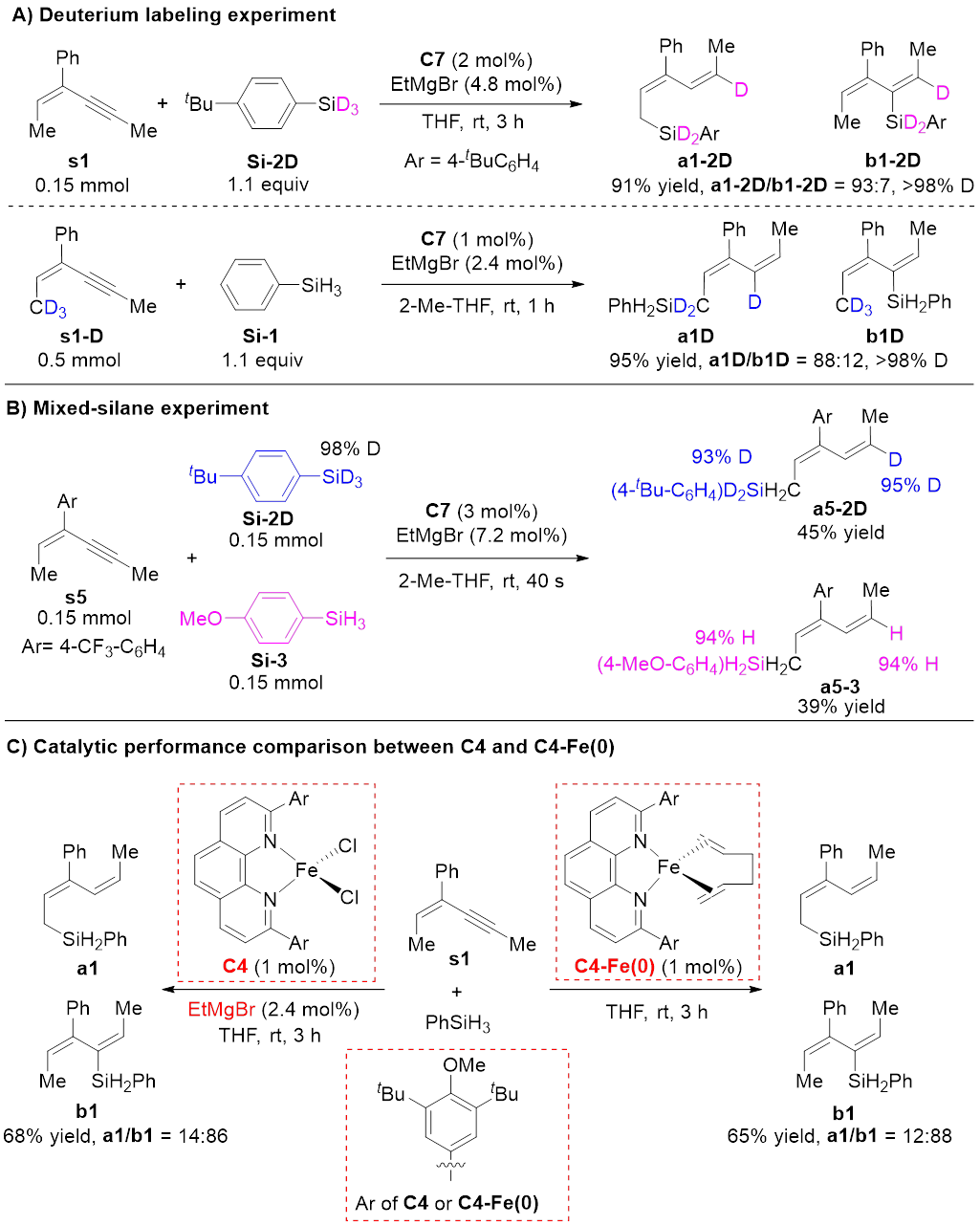
图3 控制实验
研究者提出了反应的可能机理(图4):二价铁前体催化剂在乙基格氏试剂的作用下被还原为零价铁活性物种并与共轭烯炔底物配位生成int1,int1再与硅烷配位生成int2,随后经ts1发生硅烷到炔烃的氢转移,生成烯基硅产物int3。int3可能发生两种转化,一种是直接发生还原消除生成炔烃硅氢化产物b12;另一种是发生烯丙位C(sp3)-H断裂并最终生成烯丙位C(sp3)‒H硅化产物a11。DFT计算表明,铁配合物催化剂的自旋交叉在反应中起到至关重要的作用。若烯基铁中间体生成后不发生自旋交叉,则在三重态势能面下进行C‒H键断裂(3int3–3ts2b–3int4a–3ts3),并最终生成烯丙位C(sp3)‒H键硅化产物a12;若其发生自旋交叉,则在五重态势能面下生成炔烃硅氢化产物(5int3–5ts2a–b12)。这意味着,烯基铁配合物中间体的自旋交叉与否决定了该反应的化学选择性(即C‒H键硅化产物与炔烃硅氢化),而自旋交叉效率可能是影响反应化学选择性的决定性因素。研究还发现,自旋交叉还可能在反应不同阶段加速或者阻碍反应发生:反应路径中第一个自旋交叉可以加速后续的还原消除(3ts1–MECP1–5int3–5ts2a),而第二次自旋交叉则使得后续还原消除比原自旋态势能面对应的能垒有所提升(3ts3–MECP2–5int5–5ts4)。上述两种自旋效应均未见文献报道,可能为理解自旋交叉对化学反应的影响提供参考。
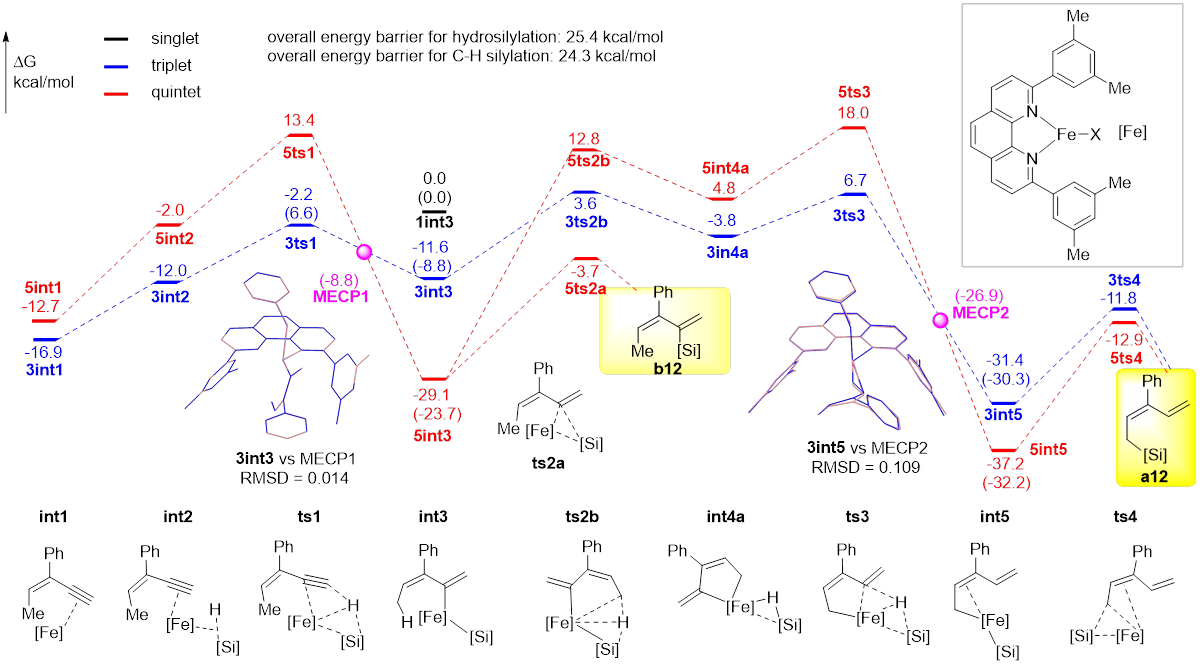
图4 可能的机理
该反应中的C‒H键活化模式与文献报道的模式都不一样,硅基不仅协助了C‒H键活化过程,显著降低了该过程的能垒,并最终转化成底物的一部分(图5)。研究者将上述C‒H键活化模式称为“底物协助的C‒H键活化”。完成氢原子转移还需经历低价铁介导的异构化过程,深入分析这一异构化过程发现其具有显著的“自旋离域调控的反应性”,即催化剂通过自旋交叉以及金属与配体间的自旋离域动态调节金属中心的自旋态和氧化态,从而加速氧化加成与还原消除这两个电性需求相反的基元反应。该过程建议的联烯中间体被合成出来,在反应条件下得到预期的C(sp3)‒H硅化的产物,支持了上述途径。
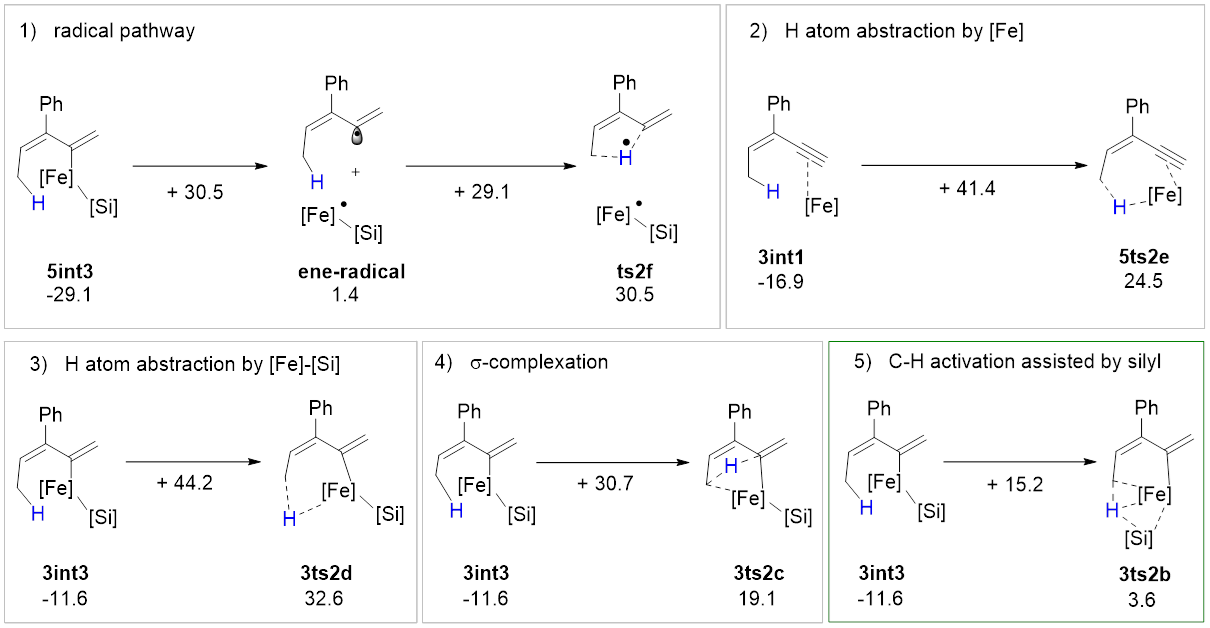
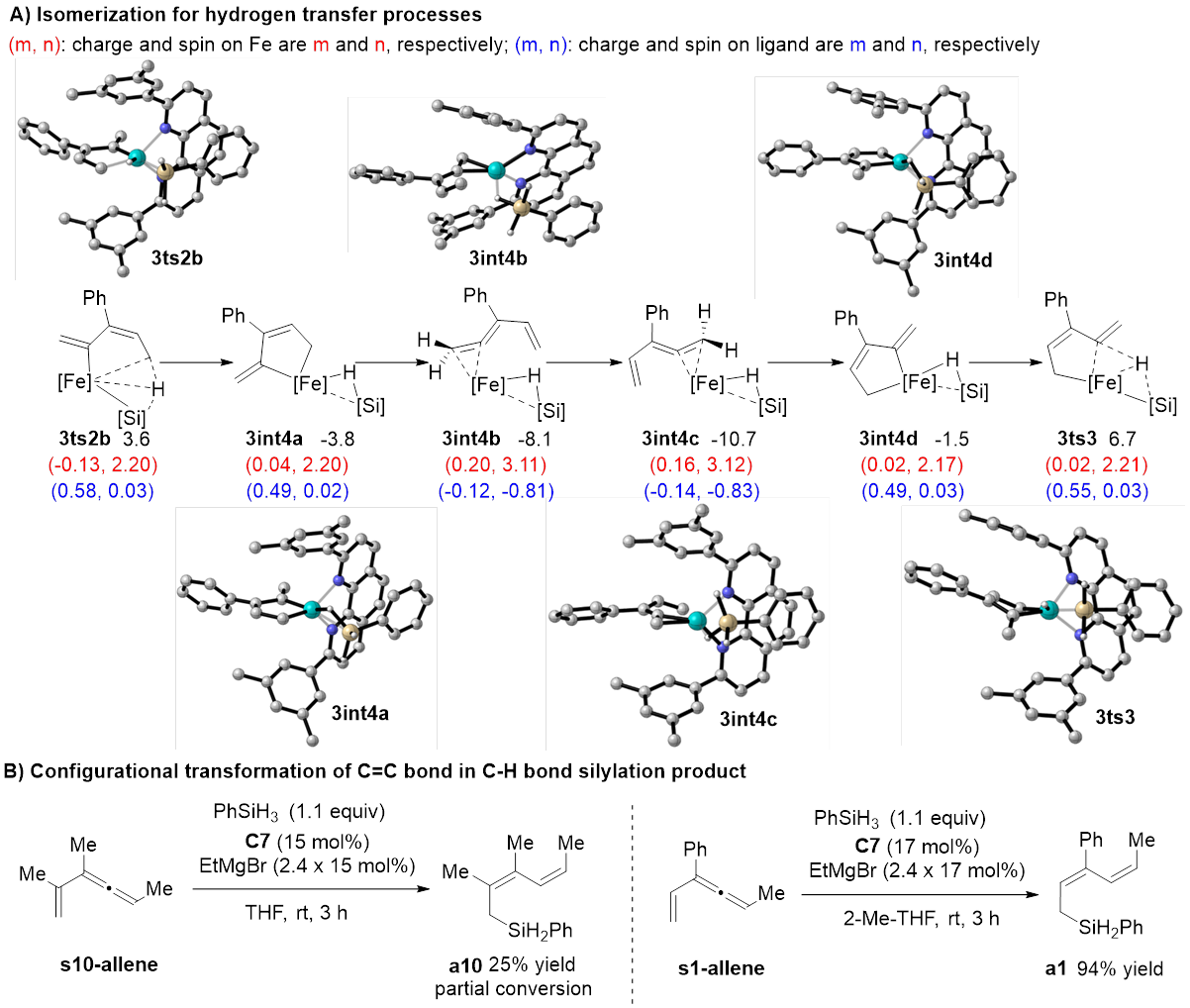
图4 C‒H键活化的可能机理
上述研究表明,铁催化剂的自旋态(特别是自旋交叉)对反应具有多重影响,不同自旋态形成的密集的势能面在新反应性发现、选择性调控等方面存在巨大的潜力,自旋有望成为调控化学反应的一个全新的维度。












No comments yet.