
本文作者:杉杉
导读
近期,美国Cornell大学T. H. Lambert教授课题组在JACS中发表论文,报道一种通过电化学光催化的Ritter型反应方法学进行的C-H键胺化研究。这一全新的C-H键胺化策略中采用TAC离子 (trisaminocyclopropenium ion) 作为催化剂,在光辐射以及电化学反应条件下,能够成功地在相应的苄位C-H键中引入乙酰氨基官能团。同时,反应过程中无需选择化学计量的各类化学氧化剂。并且,这一全新的C-H键胺化策略具有良好的官能团兼容性、广泛的底物应用范围等优势。此外,作者通过对于各类复杂分子后期衍生化过程的研究,进一步阐明这一策略的合成实用性。
C-H Amination via Electrophotocatalytic Ritter-type Reaction
T. Shen, T. H. Lambert, J. Am. Chem. Soc. 2021, 143, 8597. doi:10.1021/jacs.1c03718.

正文
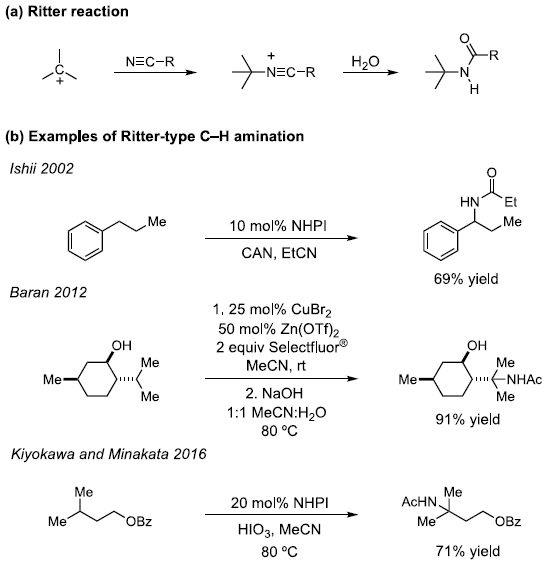
将未活化的C-H键转化为C-N键的反应策略作为有机合成领域的研究热点,一直以来备受广泛关注。其中,Hoffmann-Löfler-Freitag反应方法学已经成为构建复杂分子的关键反应之一。并且,近年来,通常采用光氧化还原催化的策略,进行Hoffmann-Löfler-Freitag反应的相关研究[1]–[3]。另一方面,通过氮烯中间体参与的一系列合成转化过程,同样能够有效地完成相应C-N键的构建。此外,采用过渡金属配合物参与的C-N偶联反应方法学,同样备受关注。同时,合成化学家进一步设计出一种新型的反应策略,将C-H键氧化,产生相应的碳正离子,并通过腈分子的捕获,最终获得一系列酰胺产物 (Figure 1a)。这一策略的本质为Ritter反应。然而,不同之处则在于碳正离子的形成方式。目前,催化Ritter型C-H胺化策略已经取得诸多的研究进展[4]–[7] (Figure 1b)。然而,却存在反应过程中选择的氧化剂与底物中的诸多官能团无法兼容的问题。由此,作者设想,设计一种无需选择化学氧化剂的电化学反应策略,则能够有效地解决上述策略中存在的官能团兼容性问题。并且,通过电化学方式实现的Ritter型C-H胺化反应方法学,同样有诸多的文献报道[8]–[11]。然而,在上述的电化学反应方案中,通常需要较高的阳极电势,因而,无法获得较高的反应收率。并且,与底物中存在的多种官能团同样无法有效地进行兼容。为进一步解决上述电化学策略中存在的相关问题,近期,本课题已经成功设计出一种通过电化学光催化条件促进的C-H双胺化 (C-H diamination)反应方法学 (Figure 1c)[12]。受上述研究的启发,这里,本文将报道一种电化学光催化的Ritter型反应方法学,进而在苄基位置引入相应的乙酰氨基官能团。同时,反应过程中无需采用化学计量的各类化学氧化剂 (Figure 1c)。
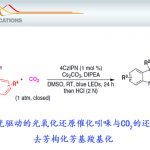
首先,作者采用2作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用8 mol% TAC 1作为催化剂,n-Bu4NPF6作为电解质,选择碳毡阳极 (carbon felt anode)与铂阴极,乙腈作为反应溶剂,同时加入TFA与水,在H型隔膜电解槽 (H-type divided cell, Ecell = 2.2 V, Eanode = 1.6 V vs SCE)以及通过CFL (compact fluorescent light)提供的可见光辐射条件下进行反应,最终获得相应的乙酰胺产物3,收率为77%。
H-type divided cell


Set up the H-type divided cell with the CFL
在获得上述最佳反应条件之后,作者开始对反应过程中的底物应用范围进行研究 (Table 2)。该小组发现,上述的标准反应条件对于甲苯与4-卤代甲苯底物,均能够以良好的反应收率,获得相应的苄基乙酰胺产物5–9。值得注意的是,该小组进一步发现,底物中卤素取代基团的位置,对于反应收率具有较为显著的影响,其中,与4-溴代产物8的反应收率相比,而2-溴代产物10与3-溴代产物11的反应收率出现显著降低。并且,反应过程中,通过高氯酸锂代替n-Bu4NPF6作为电解质,能够提高产物8与10的反应收率。同时,上述的最佳反应条件对于1-溴-3,5-二甲基苯底物,同样能够以61%的反应收率,获得相应的苄基乙酰胺产物12。
之后,作者研究发现,其它类型的烷基取代苯衍生物,同样能够顺利地参与上述的C-H键胺化过程,并获得目标产物13与14。同时,作者发现,具有支链的烷基苯底物,例如异丙苯,则最终获得相应的二氢咪唑产物15。值得注意的是,对于分子中同时具有甲基与亚甲基取代的烷基苯底物,在反应过程中,则优先选择亚甲基位点,并获得产物16,其区域异构比为6.2:1。并且,作者观察到,上述的最佳反应条件对于二苯甲烷底物,同样能够以良好的反应收率,获得相应的α-二芳基胺产物17。之后,作者进一步发现,一系列不同类型的环状底物,例如四氢萘、茚满 (indane)以及二苯并软木酮 (dibenzosuberone),同样能够顺利地参上述的转化过程,并获得产物18–20。
接下来,作者对上述C-H键胺化过程的官能团兼容性进行考察。研究表明,上述的标准反应条件对于烷基侧链中具有醇羟基、羧基、烷氧羰基以及卤素等基团取代的烷基苯底物,均能够良好地兼容,并获得相应的胺化产物21–26。同时,对于烷基侧链中具有端炔基团取代的烷基苯底物,则无法有效地进行上述的C-H键胺化过程。然而,烷基侧链中具有内炔基团取代的烷基苯底物,则能够以70%的反应收率,获得相应目标产物28。值得注意的是,上述的最佳反应条件,对于具有不同类型含氮官能团取代的芳香族化合物,同样能够有效地兼容,并获得相应的C-H键胺化产物29–34。

为进一步阐明这一全新的C-H键胺化反应策略的实用性,接下来,作者对一系列复杂有机分子进行相关的后期官能团化研究 (Table 3)。首先,该小组发现,上述的C-H键胺化策略能够有效地应用于sertraline类似物35、视黄酸受体激动剂衍生物 (retinoic acid receptor agonist derivative) 36、靛红衍生物 (isatin derivative) 37以及CYP11B1抑制剂衍生物38的后期官能团化过程,并以良好的反应收率与区域选择性,获得相应的目标产物。同时,作者观察到,具有酰亚胺以及酮羰基官能团取代的芳香族衍生物,同样能够与上述的标准反应体系良好地兼容,并获得相应的官能团化产物39–41。值得注意的是,这一全新的C-H键胺化方法学对于存在游离胺官能团的复杂分子,同样能够较好地进行兼容,并获得相应的胺化产物42–44。此外,该小组进一步发现,在通过茚满底物进行的克级规模实验研究中 (1.5 g),将反应时间延长至72 h,同样能够获得50%收率的产物41。

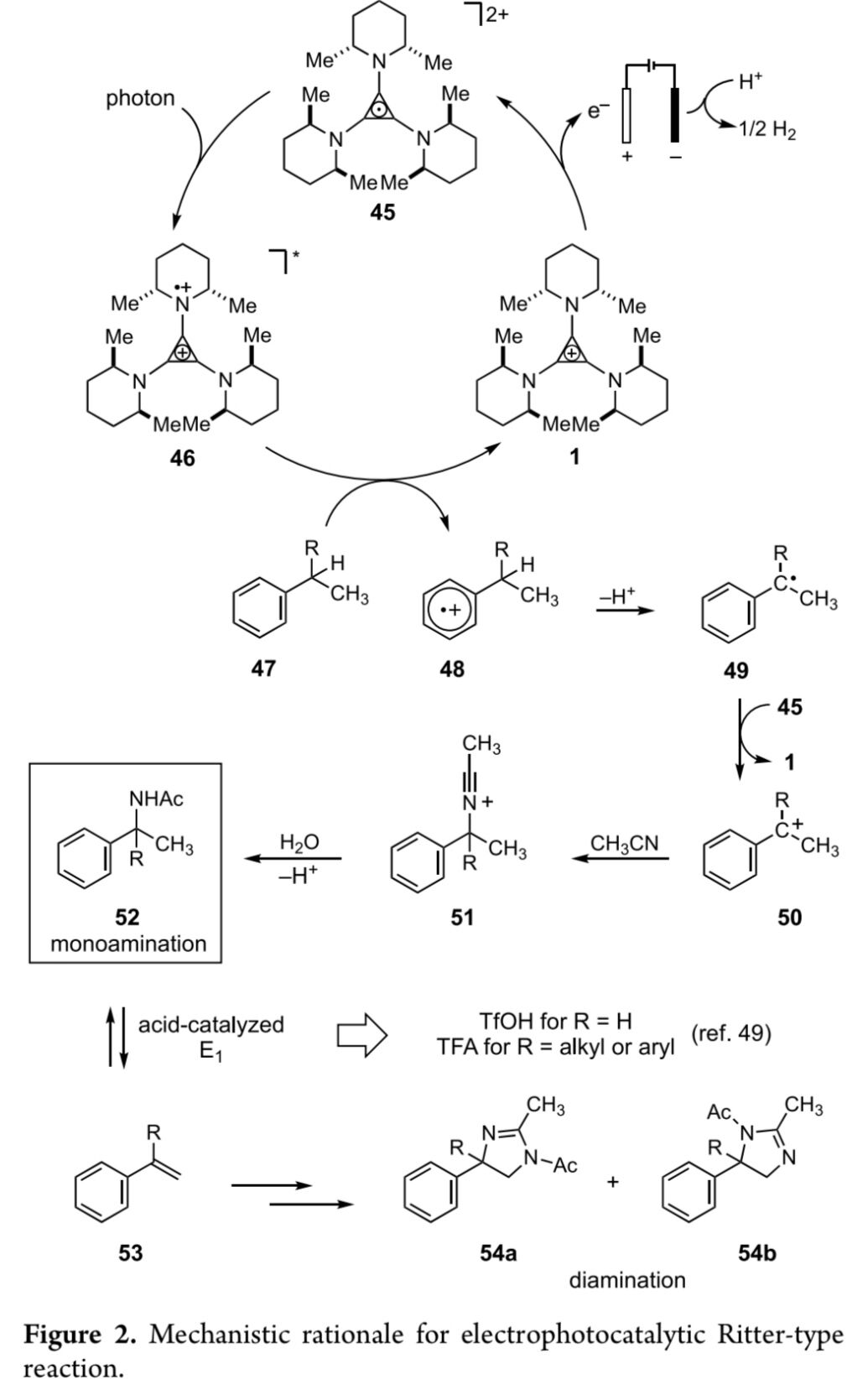
最后,作者提出一种可能的反应机理 (Figure 2)。首先,通过TAC 1的氧化过程,形成稳定的自由基双正离子45 (Ep/2 = 1.12 V in 30:1 MeCN:TFA vs SCE)。之后,45在光辐射条件下,形成光激发态中间体46,继而进一步参与芳基底物47的单电子氧化过程,产生自由基正离子48。并进一步通过48的去质子化步骤,产生苄基自由基49。接下来,苄基自由基49能够与自由基双正离子45或直接通过阳极,进行二次氧化过程,形成碳正离子中间体50。并继续通过Ritter反应路径,形成腈正离子 (nitrilium)中间体51。最终,通过51的水解过程,获得酰胺产物52。同时,通过质子的阴极还原产生氢气的反应过程,进而实现相关氧化还原过程的平衡。同时,这一电化学光催化策略设计的关键在于通过电解槽固有的电极电势,无法首先相关有机底物的氧化,因此,需要选择相应的电化学光催化剂,进而实现选择性的单电子氧化过程,并进一步减少副反应过程的发生。此外,作者进一步假设,苄基乙酰胺52经历酸催化的E1消除过程,形成苯乙烯53,之后,经历进一步的氧化与Ritter型反应过程,形成区域异构二氢咪唑54a或54b。接下来,作者进一步发现,在苄基位置存在侧链的底物中,通过TFA,能够更加容易地促进消除过程的进行。因此,在苄基位置存在侧链的底物中,并未观察到苄基乙酰胺产物52的形成。然而,在苄基位置无侧链的底物中,上述消除过程的进行,则需要选择酸性更强的三氟甲磺酸。因此,在TFA存在的反应条件下,仅能够形成苄基乙酰胺产物52。接下来,该小组通过CV (cyclic voltammetry)法对相应胺化产物的氧化还原电势进行测定 (Table 2)。通过上述的测定过程,作者发现,目标产物的氧化还原电势与起始原料极为接近(参阅Supporting Information)。并且,乙酰氨基官能团的引入,能够减慢其与TAC 1之间进行单电子氧化过程的动力学速率,因而,使上述的电化学光催化过程能够更为有效地进行。
总结
美国Cornell大学的T. H. Lambert教授课题组成功开发出一种全新的电化学光催化Ritter型反应方法学,采用乙腈作为氮源,在无相应化学氧化剂存在的条件下,成功完成一系列苄基乙酰胺分子的构建。同时,这一全新的C-H胺化策略具有良好的官能团兼容性以及广泛的底物应用范围等优势。此外,通过对各类复杂分子的后期官能团化研究,进一步阐明上述策略的合成应用价值。
参考文献
[1] E. A. Wappes, S. C. Fosu, T. C. Chopko, D. A. Nagib, Angew. Chem. Int. Ed. 2016, 55, 9974. doi: 10.1002/anie.201604704. [2] C. Martinez, K. Muñiz, Angew. Chem. Int. Ed. 2015, 54, 8287. doi: 10.1002/anie.201501122. [3] P. Becker, T. Duhamel, C. J. Stein, M. Reiher, K. Muñiz, Angew. Chem. Int. Ed. 2017, 56, 8004. doi: 10.1002/anie.201703611. [4] Q. Michaudel, D. Thevenet, P. S. Baran, J. Am. Chem. Soc. 2012, 134, 2547. doi: 10.1021/ja212020b. [5] K. Kiyokawa, K. Takemoto, S. Minakata, Chem. Commun. 2016, 52, 13082-13085. doi: 10.1039/C6CC07164C. [6] S. Sakaguchi, T. Hirabayashi, Y. Ishii, Chem. Commun.2002, 5, 516-517. doi: 10.1039/B110638D. [7] G. Li, C. A. Morales-Rivera, F. Gao, Y. Wang, G. He, P. Liu, G. Chen, Chem. Sci. 2017, 8, 7180. doi: 10.1039/C7SC02773G. [8] J. Y. Becker, L. R. Byrd, L. L. Miller, Y.-H. So, J. Am. Chem. Soc. 1975, 97, 853. doi: 10.1021/ja00837a027. [9] J. Y. Becker, L. R. Byrd, L. L. Miller, J. Am. Chem. Soc. 1974, 96, 4718. doi: 10.1021/ja00821a083. [10] J. Y. Becker, L. L. Miller, T. M. Siegel, J. Am. Chem. Soc. 1975, 97, 849. doi: 10.1021/ja00837a026. [11] T. Tajima, H. Ishii, T. Fuchigami, Electrochem.Commun. 2002, 4, 589. doi: 10.1016/S1388-2481(02)00381-8. [12] T. Shen, T. H. Lambert, Science 2021, 371, 620. doi: 10.1126/science.abf2798.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!















No comments yet.