
本文作者:杉杉
导读
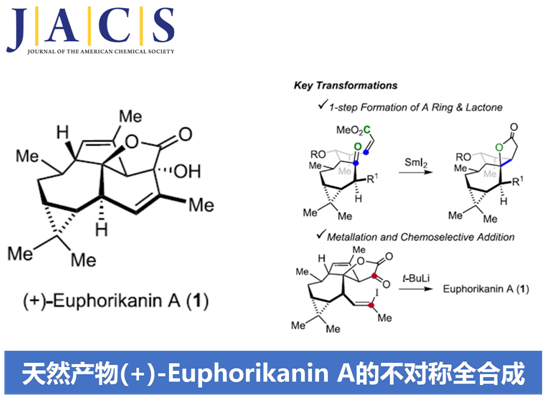
近日,Zürich联邦理工学院 (Eidgenössische Technische Hochschule Zürich, ETH Zürich)的E. M. Carreira授课题组在JACS中发表论文,首次报道了ingenane衍生的具有独特5/6/7/3稠合四环骨架 (5/6/7/3-fused tetracyclic skeleton)的天然产物(+)-Euphorikanin A的不对称全合成。其中,关键步骤涉及采用SmI2媒介促进的羰基自由基负离子-烯酸酯反应 (ketyl-enoate reaction),进而通过一步反应过程,完成两重环体系的构建。并且,这一极性反转成环的设计策略具有优良的收率以及高度的非对映选择性。同时,通过一系列化学选择性的合成转化过程 (主要涉及乙烯基锂试剂的原位产生,以及通过锂试剂对于α-酮内酯的分子内亲核进攻过程)能够在后期实现(+)-Euphorikanin A分子中B环的构建。
Enantioselective Total Synthesis of (+)-Euphorikanin A
M. J. Classen, M. N. A. Böcker, R. Roth, W. M. Amberg, E. M. Carreira,
J. Am. Chem. Soc. 2021, 143, 8261. doi: 10.1021/jacs.1c04210.
正文
Spurges (Euphorbiaceae)科植物作为生物活性天然产物的重要来源,一直以来备受关注。2016年,Zhang等[1]由Euphorbia kansui的根部,分离并鉴定出一种新型的二萜 (diterpenoid)类分子,即Euphorikanin A (1)。之后,研究发现,Euphorikanin A能够表现出相应的细胞毒性 (cytotoxicity)。并且,这一全新的二萜类分子同样具有潜在的药学研究价值。同时,Zhang提出Euphorikanin A的生物合成途径(biosynthetic pathway)中涉及ingenane的重排过程。
尽管ingenane二萜为文献报道较为充分的重要天然产物之一,然而,对于Euphorikanin A (1)全合成,则尚未有相关的文献报道。在结构特征方面,Euphorikanin A (1)具有5/6/7/3稠合的四环骨架,并带有八重的连续立体中心。同时,其分子骨架中涉及Cα-桥头中具有羟基取代的桥连[3.2.1]-γ-内酯结构单元。
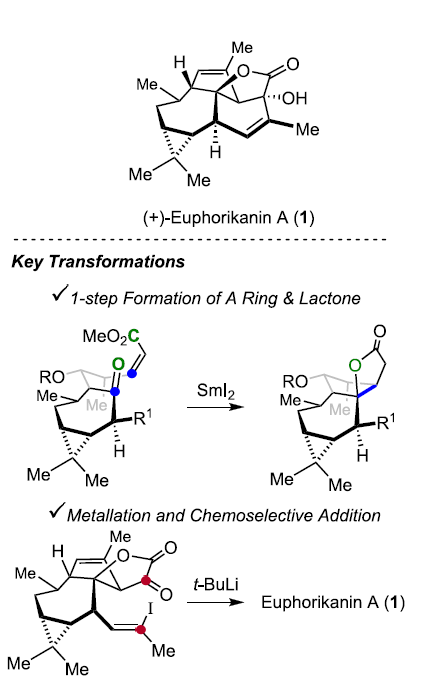
这里,本文首次报道采用(+)-3-蒈烯 ((+)-3-carene) 2作为起始原料,进而成功实现(+)-Euphorikanin A (1)分子的不对称全合成设计。这一全合成设计方案中的关键步骤主要涉及羰基自由基负离子-烯酸酯环化反应 (ketyl-enoate)以及有机锂物种对于α-酮内酯 (α-ketolactone)的化学选择性分子内加成过程 (Scheme 1)。
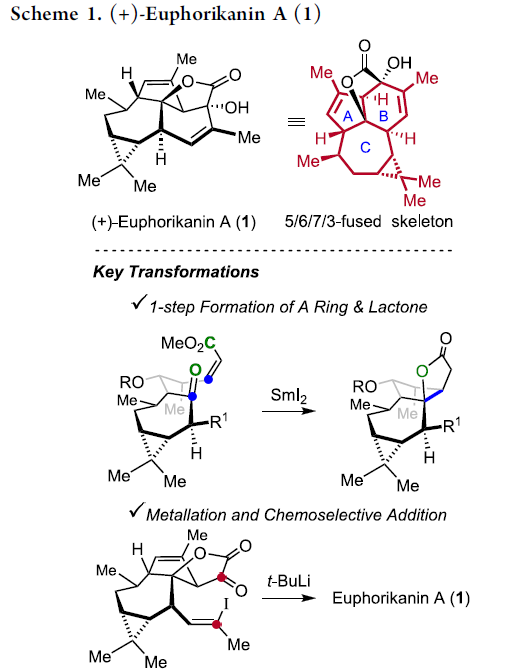
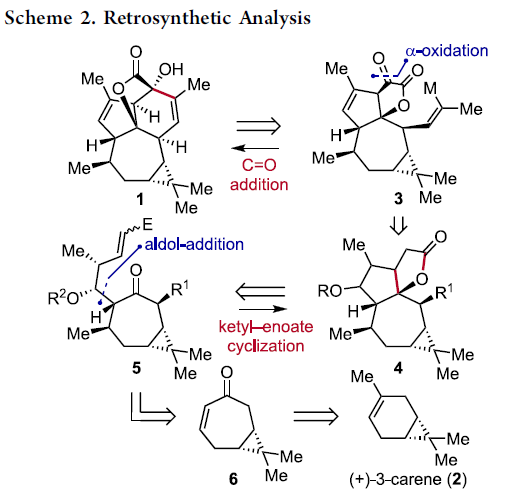
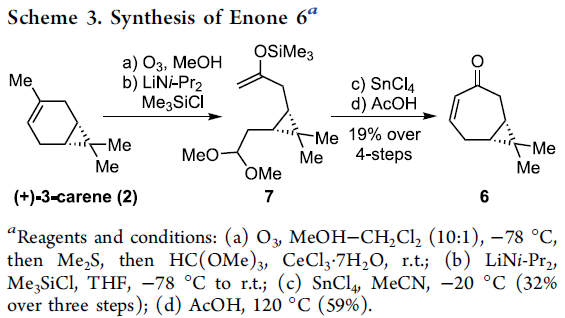
该小组通过六元环B的构建开始,进行相关的逆合成设计 (Scheme 2, 1 ⇒ 3)。首先,作者设想,通过关键砌块3的分子内羰基加成步骤,最终能够实现 (+)-Euphorikanin A (1)的全合成过程。其中,砌块3能够通过砌块4的α-氧化过程而获得。而砌块4中较为关键的稠合[5,5]双环骨架能够通过极性反转的串联环化策略的设计,即通过化合物5参与的羰基自由基负离子-烯酸酯反应[2]-[5]而顺利实现。同时,化合物5能够通过简单的环庚烯酮6进行制备。而环庚烯酮6则能够进一步通过更为廉价易得的(+)-3-蒈烯原料 (2),通过手性池 (chiral pool)策略,较为简便地进行制备[6]。
前期,Yamakawa团队[6]报道采用(+)-3-蒈烯 (2)作为起始原料,顺利完成环庚烯酮6的制备。这里,作者对上述反应条件进行进一步的改进,进而获得较大量的环庚烯酮6 (Scheme 3)。首先,将(+)-3-蒈烯(2)在-78 oC条件下进行臭氧分解,并通过后续的CeCl3·7H2O处理,获得二甲基缩醛。之后,在动力学控制的反应条件下,采用LDA与Me3SiCl进行处理,形成烯醇醚7。接下来,在SnCl4存在的条件下,烯醇醚7经历三步的反应过程,并获得32%收率的Mukaiyama羟醛缩合产物。之后,通过在乙酸媒介中的加热回流过程,最终完成环庚烯酮6的制备。上述的反应过程仅需要通过两次快速硅胶柱色谱的分离纯化操作,即可获得较大量的环庚烯酮产物6。
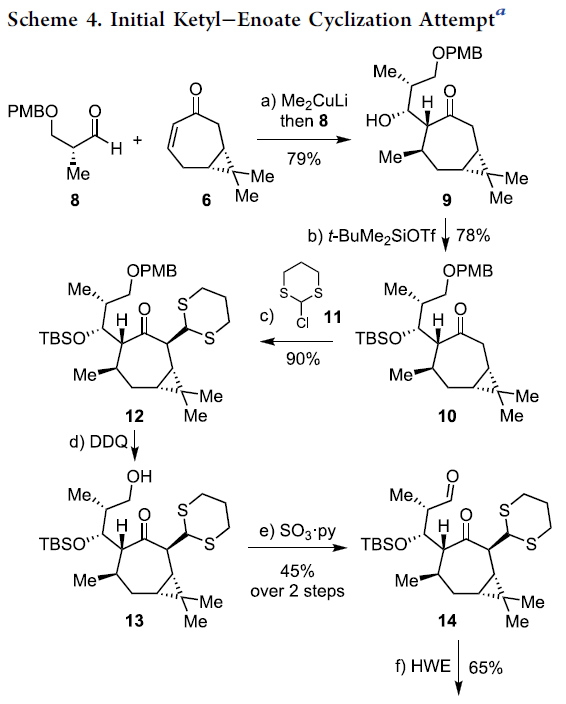
环庚烯酮6通过一锅共轭加成 (与Me2CuLi试剂)/羟醛加成 (与醛8)的反应序列,进而以79%的分离收率,获得单一的非对映体二级醇产物9 (Scheme 4)。之后,二级醇产物9通过硅基化步骤,获得78%收率的产物10。接下来,产物10通过LiN(SiMe3)2与氯代二噻烷11参与的烷基化过程,以90%的收率获得单一的非对映体12。
通过DDQ对PMB-醚12的氧化裂解过程,能够获得一级醇13。之后,将未分离纯化的一级醇13通过Parikh-Doering氧化步骤,并以45%的收率获得相应的醛14。接下来,将14 通过Horner-Wadsworth-Emmons烯基化反应步骤,形成相应的E-烯酸酯15。
15在THF溶剂中,通过SmI2与2eq. MeOH 处理,获得单一手性产物16,收率为78%。接下来,作者假设,存在两种较为关键的因素,进而有效地实现反应过程中立体选择性的控制:首先,通过酮以及酯基团与钐中心的螯合过程,形成环状过渡态。同时,环状过渡态中,不同基团之间采取立体相互作用最小化的反应活性构象。通过上述两种因素的共同作用,能够使相应的羰基自由基负离子-烯酸酯环化过程表现出优良的立体选择性。
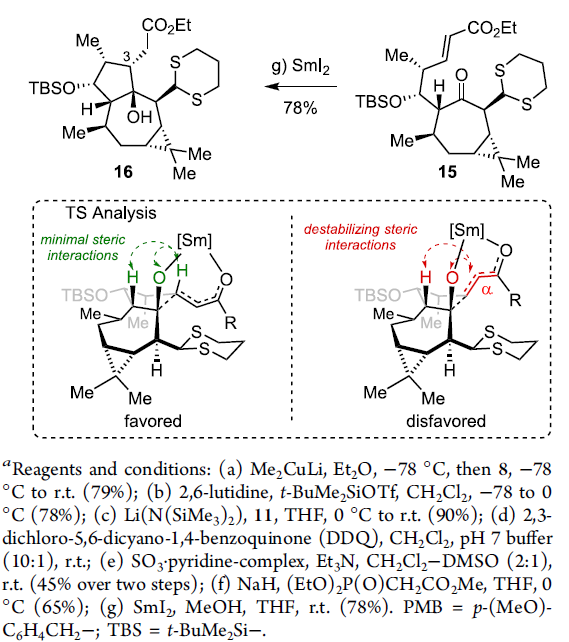
通过Scheme 4中的分析,作者进一步设想采用Z-烯酸酯参与的羰基自由基负离子-烯酸酯环化过程,将获得相反的C(3)立体选择性。其中,Tori等[7]研究表明,烯酸酯的几何构型对于简单底物的相关环化过程具有较为显著的影响。接下来,作者通过醛14的Still-Gennari烯基化反应,获得76%收率的Z-烯酸酯化合物17 (Scheme 5)。之后,研究发现,化合物17采用上述SmI2媒介的环化方案,能够以优良的收率,获得相应的γ-内酯化合物18。

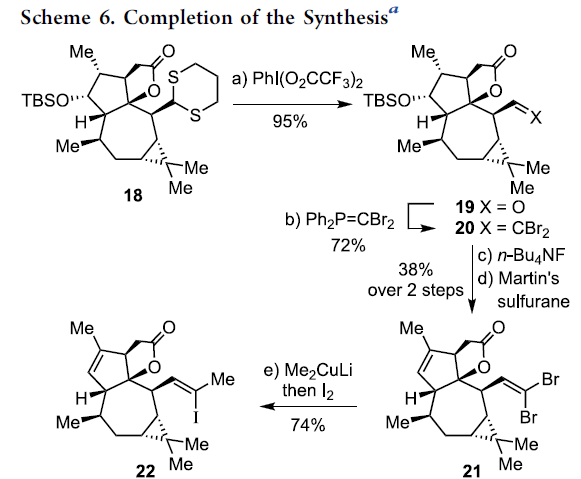
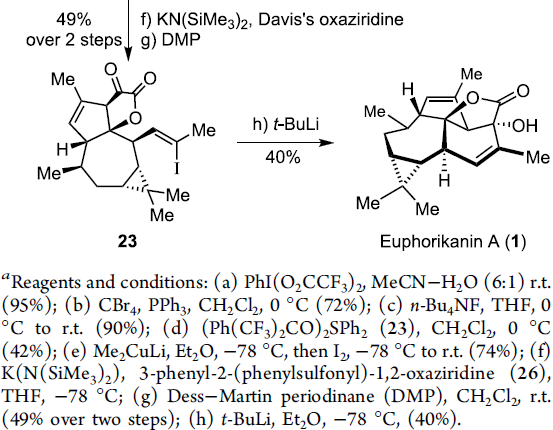
在通过上述的设计方案,研究成功完成(+)-Euphorikanin A中两重环骨架的构建。接下来,后续反应方案的设计中,面临的全新挑战则主要涉及B环的构建以及A环中双键的引入 (Scheme 6)。首先,作者将化合物18在乙腈-水混合溶剂中,采用PIFA进行处理,进而去除分子中的二噻烷结构,并获得95%收率的醛19,之后,19经历Ramirez烯基化步骤,形成1,1-二溴代烯20。1,1-二溴-烯烃20通过n-Bu4NF的去保护以及后续的Martins sulfurane处理,将获得砌块21及其区域异构体。接下来,作者选择Tanino、Miyashita等[8]-[9]报道的合成转化策略,即将21采用Me2CuLi与I2进行处理,获得乙烯基碘砌块22。接下来,将22采用Davis oxaziridine进行处理,进而完成相应的α-羟基化过程。之后,再通过Dess-Martin氧化过程,获得α-酮内酯23。最终,该小组发现,在-78oC以及Et2O溶剂中,采用t-BuLi处理,能够将23中的乙烯基碘结构单元转化为相应的乙烯基锂试剂,并进一步通过有机锂中间体对α-酮内酯的化学选择性的分子内亲核进攻过程,进而以40%的反应收率,成功完成(+)-Euphorikanin A (1)的不对称全合成。同时,作者进一步观察到,实验合成出的(+)-Euphorikanin A (1)产物的全部表征数据 (主要涉及1H-NMR、13C-NMR、HRMS、IR以及[α]D)均能够与前期文献报道的相关数据良好地匹配[1]。
总结
Zürich联邦理工学院的E. M. Carreira授课题组首次报道(+)-Euphorikanin A的不对称全合成。其中,作者采用(+)-3-蒈烯 (2)作为起始原料,通过19步的反应过程,成功完成天然产物(+)-Euphorikanin A的对映选择性构建。该小组在相关全合成方案的设计中,采用氯代二噻烷11作为有效的甲酰基正离子等价物 (formyl cation equivalent)。同时,通过SmI2媒介促进的羰基自由基负离子-烯酸酯环化串联反应的关键步骤,能够一步实现A环以及γ-内酯环的构建。之后,作者采用α-酮内酯的化学选择性加成策略,最终完成(+)-Euphorikanin A分子中B环的构建。















No comments yet.