
本文作者:Joy
导读
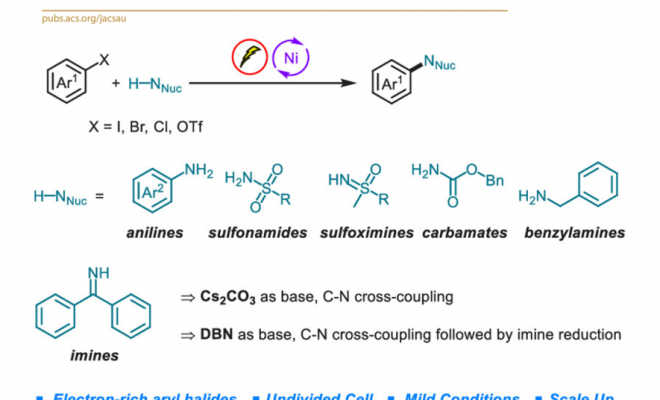
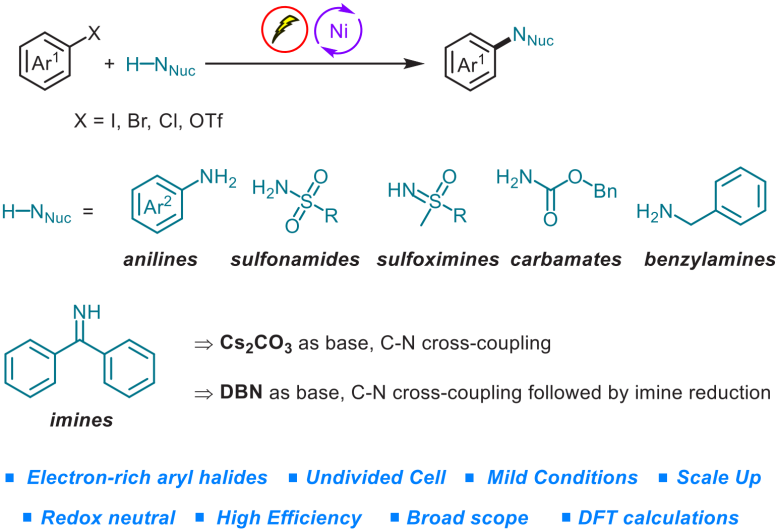
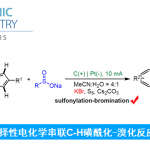
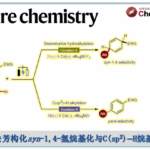
Abdullah国王科技大学 (King Abdullah University of Science and Technology, KAUST) 的M. Rueping与H. Yue团队成功实现在镍电催化(nickelaelectrocatalysis)条件下,采用弱N-亲核试剂(weak N-nucleophile)参与的交叉偶联胺化反应方法学。研究表明,芳卤与芳基对甲苯磺酸酯能够通过协同成对电解 (concerted paired electrolysis)的方式与一系列弱N-亲核试剂进行有效的偶联,弱N-亲核试剂主要涉及苯胺、磺酰胺、亚砜亚胺 (sulfoximine)、氨基甲酸酯以及亚胺。值得注意的是,缺电子苯胺与磺酰胺类底物同样能够适用于上述的偶联反应。更为有趣的是,将二苯甲酮亚胺应用于上述偶联胺化过程时,可以通过采用不同类型的碱,选择性地获得胺或亚胺产物。除此之外,作者发现采用交流电模式 (alternating current mode),同样能够有效地完成上述偶联胺化反应。
Redox-Neutral Cross-Coupling Amination with Weak N-Nucleophiles: Arylation of Anilines, Sulfonamides, Sulfoximines, Carbamates, and Imines via Nickelaelectrocatalysis
C.Zhu, A.P. Kale, H. Yue, M. Rueping, JACS Au 2021, doi: 10.1021/jacsau.1c00148.
正文
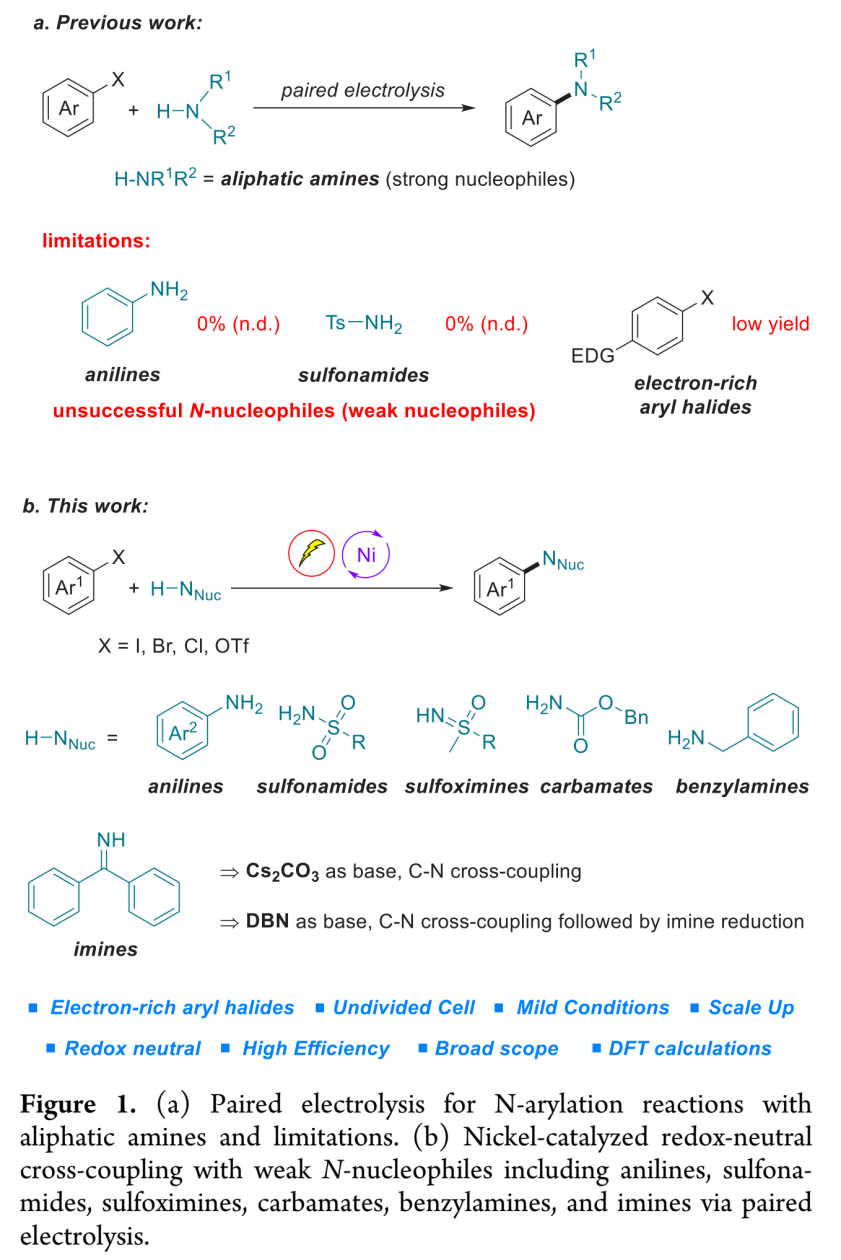
含氮有机分子已经成为有机化合物中最为重要的类别,能够广泛应用于合成、制药以及材料科学等领域。因此,合成化学家一直致力于发展一种构建C-N键的通用且有效的方法。通过钯、铜或镍催化[1]-[4] 的有机卤代物的胺化反应方法学进行C-N键的构建,目前已经取得较大的研究进展,然而,这一方法学仍然存在较多的不足之处,例如,反应过程中需要选择空气敏感或价格昂贵的金属催化剂,需要加入复杂的配体,采用强碱或较高的反应温度。上述问题致使该方法学的底物应用范围以及该方法学在不同研究领域中的进一步应用受到较大的限制。
为解决上述问题,目前已经开发出双重催化策略 (dual catalytic strategy) [5]-[8],这一策略能够在温和的反应条件下完成一系列胺分子的构建。由于有机电化学 (organic electrochemistry)具有能源经济性 (energy economy)、可持续性 (sustainability)、反应条件温和 (mild reaction condition)、可调节性 (adjustability)与可放大性 (scalability)等优势[9],近年来备受有机化学家的广泛关注。其中,成对电解 (paired electrolysis)由于其阳极与阴极中的半反应能够同时用于形成所需的中间体或产物[10],因而成为有机合成化学家关注的热点。更为重要的是,将电解过程与镍催化有效地结合,能够在温和的反应条件下完成C-C键与 C-杂原子键的构建。在此背景下,Baran课题组率先开发出镍电催化的芳卤以及三氟甲磺酸酯与脂肪胺之间的胺化反应,并将底物应用范围扩展至氨基酸酯、核苷 (nucleoside)与寡肽 (oligopeptide)[11]。然而,这一先导性的研究工作中,相关的反应条件无法有效地应用于富电子的芳卤以及弱亲核性的苯胺与磺酰胺化合物 (Figure 1a)。基于胺类化合物在化学研究领域中的重要价值以及目前交叉偶联反应方法学的局限性,作者致力于发展一种更加通用的胺化反应方案。在此,作者报道一种在镍电催化条件下,芳基亲电底物与一系列弱亲核试剂 (主要涉及苯胺、磺酰胺、亚砜亚胺、氨基甲酸酯、苄胺以及亚胺)通过成对电解 (paired electrolysis)方式,形成C-N键的交叉偶联胺化反应方法学 (Figure 1b)。
(图片来源:JACS Au)
首先,作者采用4-溴苯腈 1a与对甲苯胺 2a之间的交叉偶联作为模板反应,通过对电流、镍催化剂、配体、电解质以及碱等相关条件进行筛选,确定最佳的反应条件为 (Table1):采用NiCl2·dme作为催化剂,dtbbpy (4,4′-di-tert-butyl-2,2′-dipyridyl)作为配体,Et3N作为碱,nBu4NBr作为支持电解质,DMA作为反应溶剂,采用石墨/泡沫镍 (graphite/nickel foam)作为电极,在无隔膜电解槽 (undivided cell)中,室温条件下、并调节电流强度为4 mA,最终能够以72%的分离产率获得相应目标产物3a。

(图片来源:JACS Au)
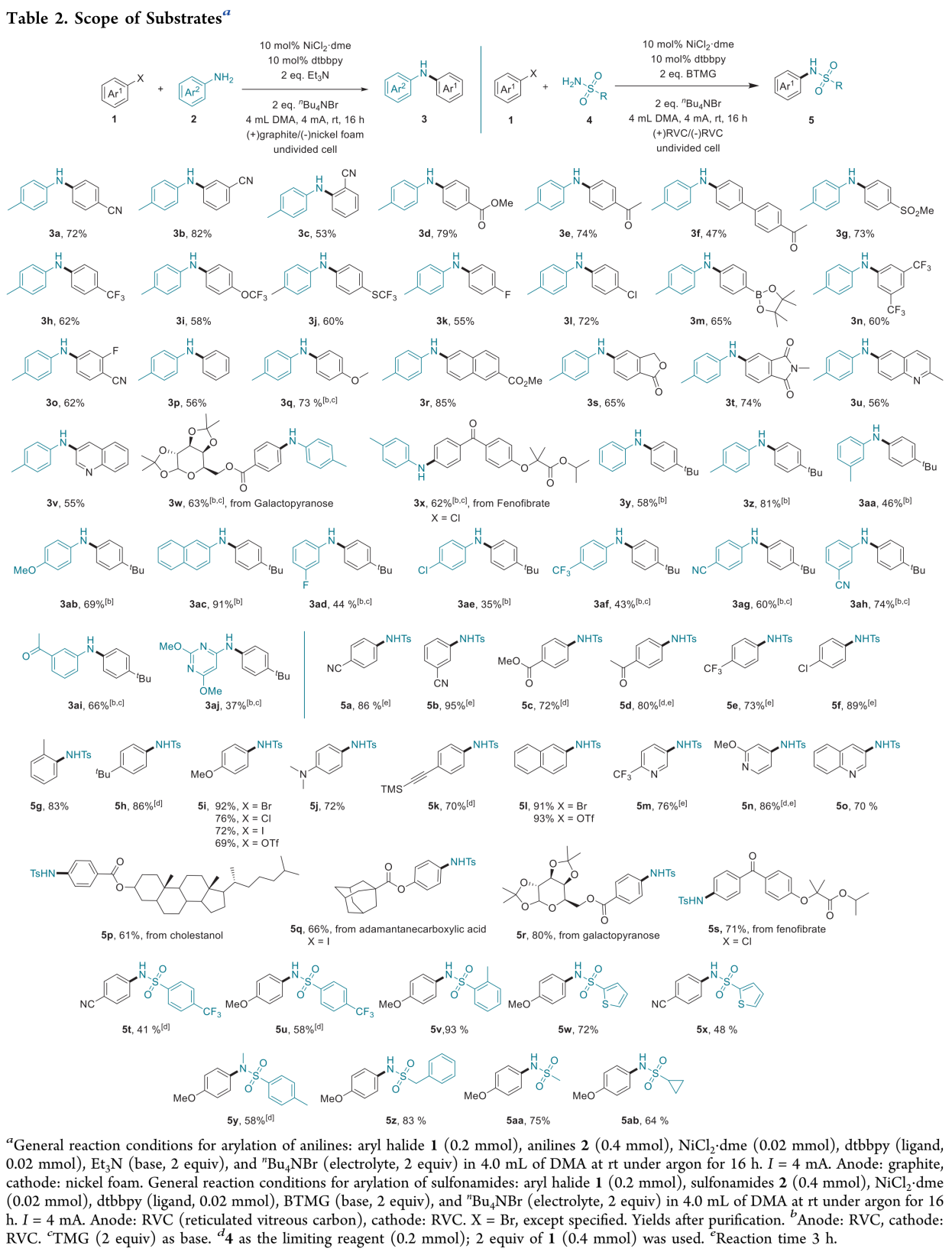
在上述的最佳反应条件下,作者对芳基溴与苯胺的底物范围进行考察 (Table 2)。研究表明,一系列具有不同电子效应与立体效应的芳基溴底物均能够与上述镍电催化的反应条件有效地兼容,并获得良好的产物分离收率。并且,上述反应过程具有优良的化学选择性,对于带有氰基、酯基、酮羰基、砜、三氟甲基、三氟甲氧基、三氟甲硫基取代的芳基溴底物 (3a–3j)、具有显著立体位阻的邻位取代芳基溴 (3c)、带有氟、氯或硼酸酯等反应活性官能团取代的芳基溴 (3k–3m)、二取代芳基溴 (3n–3o) 以及中性与富电子的芳基溴 (3p–3q)均能够良好地兼容。此外,作者发现,具有内酯、酰亚胺与喹啉结构单元的双环底物 (3s–3v)以及吡喃半乳糖衍生的芳基溴与fenofibrate (3w–3x)同样能够有效地参与上述交叉偶联胺化过程,进而表明这一全新的交叉偶联胺化方法学在医药相关化合物 (pharmaceutical-related compound)的官能团化研究中具有良好的应用潜力。同时,该小组对于苯胺底物应用范围的研究表明,一系列缺电子、富电子以及电中性的苯胺底物,均能够有效地参与上述偶联胺化过程 (3y-3ac),并获得良好的收率。同时,作者进一步观察到,具有较弱亲核性的N-亲核试剂,例如带有氟、氯、三氟甲基、氰基以及乙酰基取代的苯胺底物,能够获得中等至良好收率的偶联胺化产物 (3ad-3ai)。而且,研究发现,杂环氨基化合物,例如氨基嘧啶同样能够有效地进行上述偶联胺化过程 (3aj)。接下来,作者对其它类型的N-亲核试剂进行进一步考察。研究表明,通过适当调整反应装置与部分反应试剂,同样能够实现一系列芳卤化合物的磺酰胺化。该小组在改用(+)RVC (reticulated vitreous carbon)/(−)RVC电极,同时改用BTMG (2-tert-butyl-1,1,3,3-tetramethylguanidine)作为碱的条件下,对芳基亲电底物与磺酰胺类化合物之间交叉偶联磺酰胺化过程的底物适用范围进行深入研究。实验发现,相应的磺酰胺化过程同样具有广泛的底物应用范围。同时,磺酰胺化过程的反应条件对于一系列带有不同官能团取代的富电子或缺电子的芳基溴均能够良好的兼容,并获得良好至优良的反应收率 (5a–5i)。值得注意的是,磺酰胺化过程的反应条件对于炔基取代的芳基溴 (5k)以及具有显著立体位阻的2-溴甲苯 (5g)同样能够良好地兼容,这表明立体位阻对于磺酰胺化 (sulfonamidation)过程无显著影响。此外,作者通过进一步实验发现,芳基溴、芳基碘、芳基氯以及芳基对甲苯磺酸酯均能够有效地完成相应的磺酰胺化反应 (5i 与 5l)。更为重要的是,一系列药物相关的带有吡啶、喹诺酮 (quinolone)以及噻唑骨架的杂环底物与天然产物衍生的复杂分子 (natural-product-derived complex molecule)同样能够有效地参与上述的偶联磺酰胺化过程,进而获得一系列具有结构多样性的生物活性化合物 (5m–5s)。同时,实验过程中,作者同样观察到,炔电子的芳基磺酰胺 (5t与5u)、邻位甲基取代的芳基磺酰胺(5v)、二级芳基磺酰胺与噻吩-2-磺酰胺 (5w-5y)以及脂肪族磺酰胺 (5z-5ab)同样表现出良好的反应活性。
(图片来源:JACS Au)
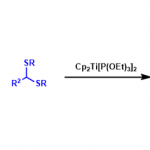
接下来,作者进一步完成苯胺与磺酰胺的N-芳基化反应的克级规模放大。值得注意的是,磺酰胺的放大规模反应能够在2 h内完成,进而表明这一胺化反应方法学的合成有效性与实用性 (Figure 2a)。此外,作者进一步观察到,采用交流电流 (alternating current) (正弦波 (sine wave), ±3 V, 2 Hz,Figure 2b),同样能够有效地完成上述的磺酰胺化反应。此外,作者顺利将上述镍电催化胺化反应方法学应用于其它不同类型的N-亲核试剂,例如亚砜亚胺 (sulfoximines)、苄胺以及氨基甲酸酯,并获得良好的反应收率 (6a–6e,Figure 2c)。
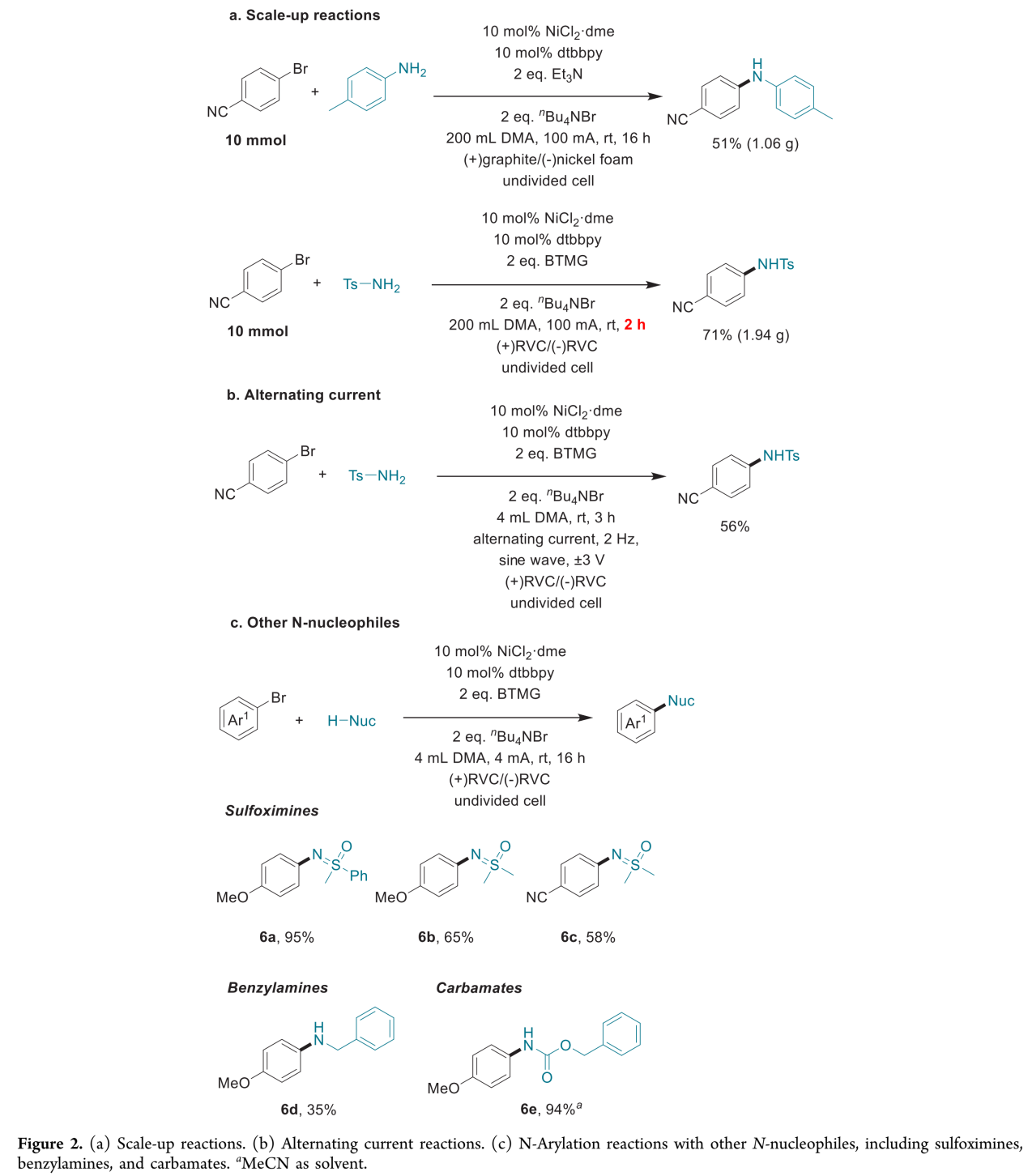
(图片来源:JACS Au)
有趣的是,作者发现,在采用二苯甲酮亚胺参与上述偶联反应时,只需要简单地调整碱的种类,便能够选择性地获得亚胺或胺产物 (Figure 3)。在采用Cs2CO3作为碱时,上述的C-N交叉偶联反应能够获得亚胺产物,并通过后续的水解过程,最终获得伯胺产物 (6f与6g)。然而,在采用有机碱DBN (1,5-diazabicyclo[4.3.0]non-5-ene)时,则首先产生亚胺产物,随后,发生进一步的还原,获得较高收率的二苯甲基保护的胺产物 (benzhydryl-protected amine) (6h)。接下来,该小组选择在d7-DMF溶剂中进行相应的控制实验,最终未观察到含氘产物 (D-incorporated product)的形成,这一事实表明氢源并非源自于反应溶剂。同时,作者研究发现,在采用DBN作为碱的电化学反应条件下,无论镍催化剂与dtbbpy配体是否存在,亚胺 6f均能够完全转化为胺 6h。这一结果表明,胺的产生源自于亚胺,有机碱可能作为相应的氢源 (还原剂)。
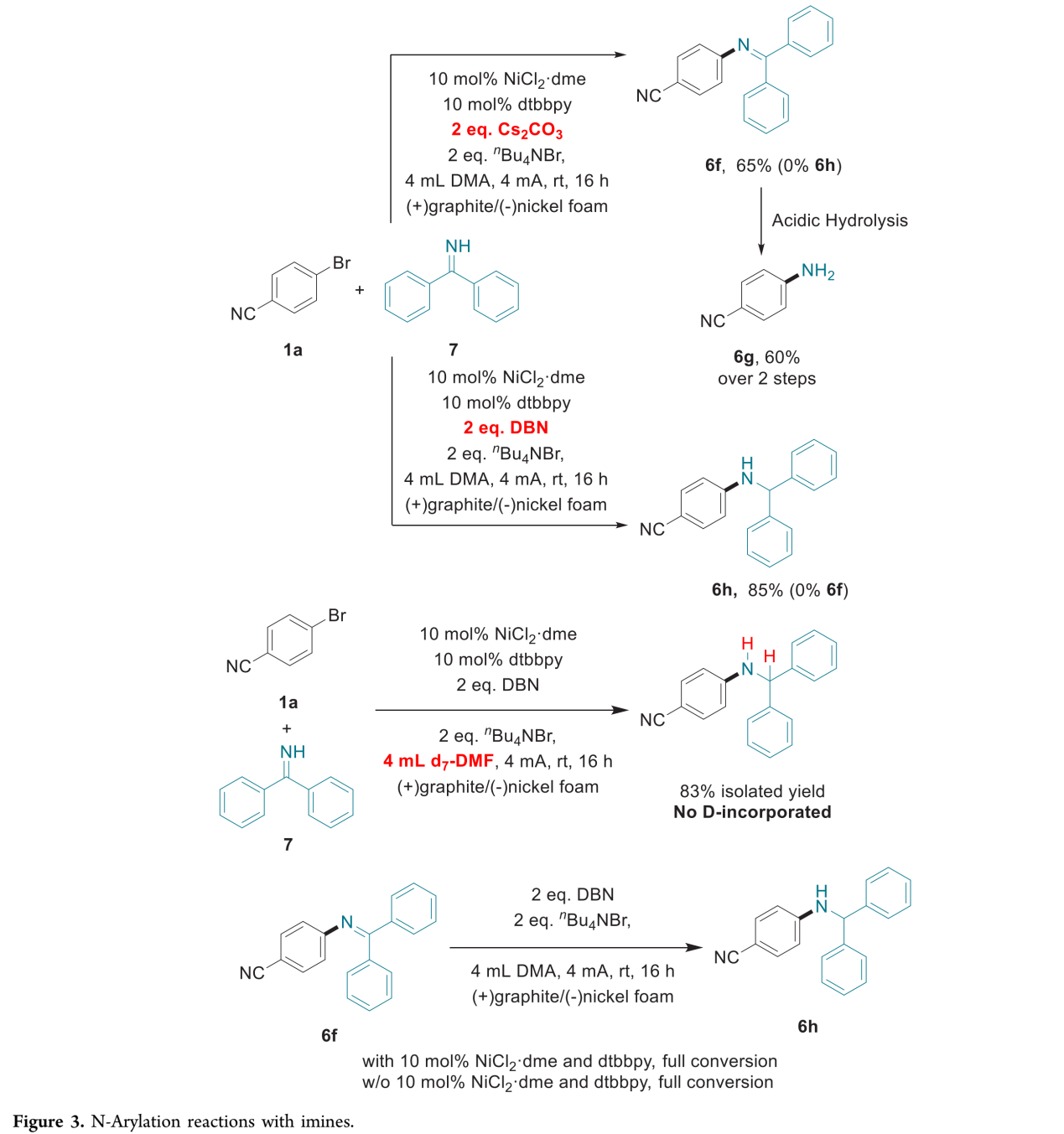
(图片来源:JACS Au)
而且,作者发现,将磺酰胺化反应在隔膜电解槽 (divided cell)中进行时,并未观察到相应目标产物的生成,这一事实表明,磺酰胺化过程为成对电解,阳极与阴极对目标产物的产生均具有重要作用 (Figure 4a)。
接下来,该小组通过对反应过程的监控,进一步发现,在采用4 mA的电流强度时,相应的反应过程能够在50 min内完成 (GC产率84%),进而表明这一方法学的合成有效性。值得注意的是,上述反应在40 min时的电流效率 (current efficiency)为154%,这表明电流的作用是抑制不稳定的NiI与NiIII中间体之间的逆歧化反应 (comproportionation) (Figure 4b)。此外,作者进一观察到,将4-溴苯腈与TsNH2在无外加电流存在的条件下,采用Ni(cod)2作为催化剂参与反应时,反应过程的有效性显著降低,这表明NiII中间体的还原消除过程较难进行 (Figure 4c),这一假设通过DFT计算获得进一步证实。DFT计算表明,胺化反应中NiII与NiIII配合物的还原消除能垒分别为26.1 kcal/mol与6.7 kcal/mol,而磺酰胺化反应中还原消除的能垒分别为34.2 kcal/mol与13.2 kcal /mol (Figure 4d)。上述事实表明,在胺化过程中,NiIII配合物更容易进行后续的还原消除过程。
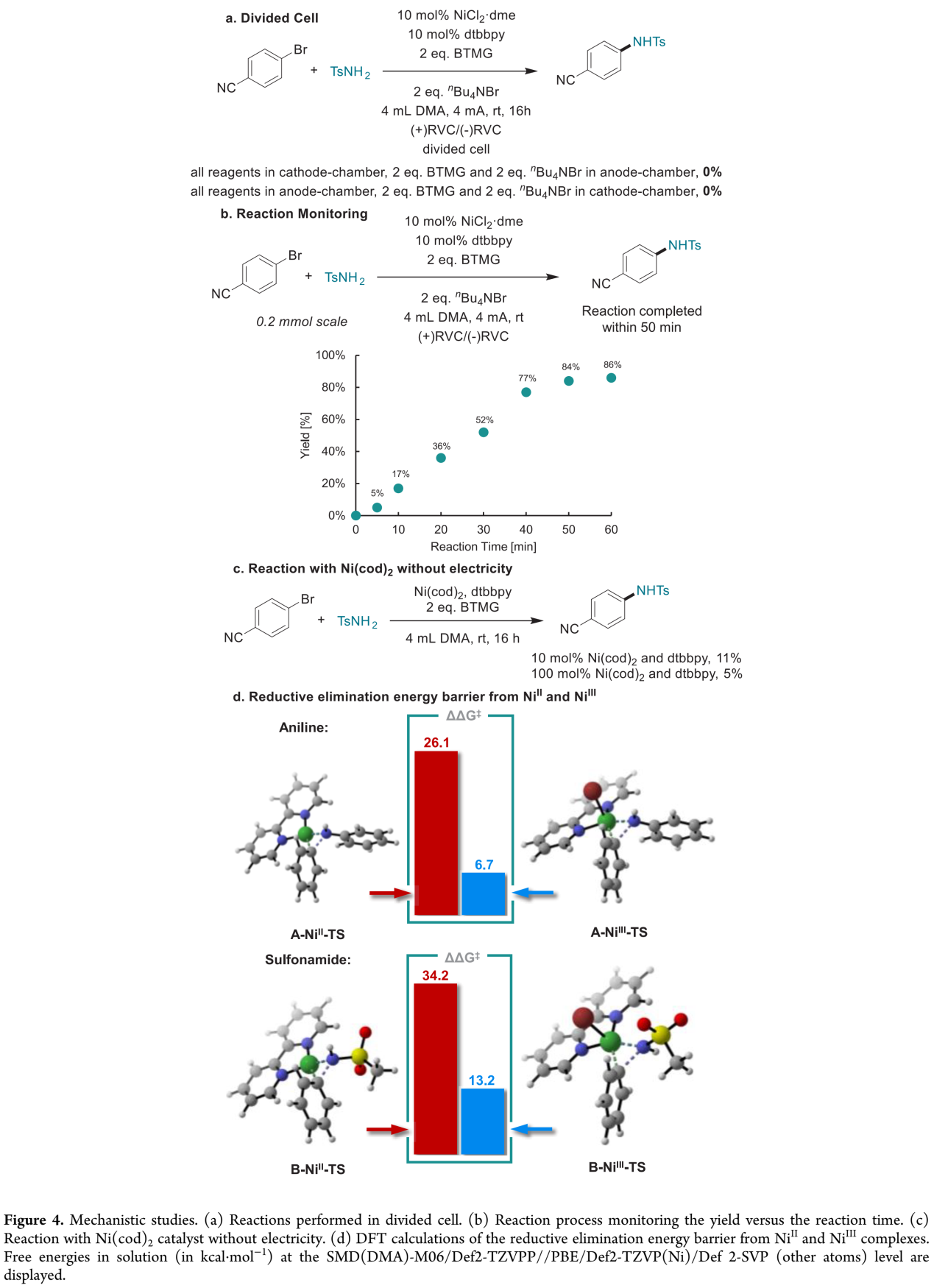
(图片来源:JACS Au)
总结
Abdullah国王科技大学的M. Rueping与H. Yue团队开发出一种高效并通用的镍电催化体系,用于形成不同类型的C-N键。在温和的电化学反应条件下,一系列不同类型的弱N-亲核试剂,例如苯胺、磺酰胺、氨基甲酸酯以及亚砜亚胺与亚胺均能够高效地参与相应的偶联胺化反应。通过缺电子苯胺与药物相关杂环分子以及复杂分子在上述偶联转化过程中的成功应用,进一步证实这一全新的偶联胺化方法学具有良好的应用价值。此外,该方法学同样能够成功实现克级规模的放大。同时,能够通过选择不同类型的碱,有效地调控形成亚胺与胺的选择性。并且,研究发现,采用交流电模式,同样能够有效地完成相关的偶联过程。最后,作者通过DFT计算,证实NiIII中间体更容易进行后续的还原消除过程,从而使这一偶联胺化反应能够在温和的反应条件下进行。综上所述,这一全新的通过镍电催化的氧化还原中性偶联胺化方法学为一系列含氮有机分子的构建开辟出一种与其他方法学有效互补、底物应用范围广泛并且更加高效适用的途径。
参考文献
(1) J. P. Tassone, E. V. England, P. M. Mac Queen, M. J. Ferguson, M. Stradiotto, Angew. Chem, Int. Ed. 2019, 58, 2485. doi: 10.1002/anie.201812862.
(2) R. T. Mc Guire, C. M. Simon, A. A. Yadav, M. J. Ferguson, M. Stradiotto, Angew. Chem. Int. Ed. 2020, 59, 8952. doi: 10.1002/anie.202002392.
(3) D. Han, S. Li, S. Xia, M. Su, J. Jin, Chem. – Eur. J. 2020, 26, 12349. doi: 10.1002/chem.202002800.
(4) R. Sun, Y. Qin, D. G. Nocera, Angew. Chem., Int. Ed. 2020, 59, 9527. doi: 10.1002/anie.201916398.
(5) C. Zhu, H. Yue, J. Jia, M. Rueping, Angew. Chem. Int. Ed. 2021, Early View. doi: 10.1002/anie.202013852.
(6) E. B. Corcoran, M. T. Pirnot, S. Lin, S. D. Dreher, D. A. Di Rocco, I. W. Davies, S. L. Buchwald, D. W. MacMillan, Science 2016, 353, 279. doi: 10.1126/science.aag0209.
(7) M. S. Oderinde, N. H. Jones, A. Juneau, M. Frenette, B. Aquila, S. Tentarelli, D. W. Robbins, J. W. Johannes, Angew. Chem. Int. Ed. 2016, 55, 13219. doi: 10.1002/anie.201604429.
(8) M. O. Konev, T. A. Mc Teague, J. W. Johannes, ACS Catal. 2018, 8, 9120. doi: 10.1021/acscatal.8b02954.
(9) Y. Yuan, A. Lei, Acc. Chem. Res. 2019, 52, 3309. doi: 10.1021/acs.accounts.9b00512.
(10) T. Wu, B. H. Nguyen, M. C. Daugherty, K. D. Moeller, Angew. Chem. Int. Ed. 2019, 58, 3562. doi: 10.1002/anie.201900343.
(11) Y. Kawamata, J. C. Vantourout, D. P. Hickey, P. Bai, L. Chen, Q. Hou, W. Qiao, K. Barman, M. A. Edwards, A. F. Garrido-Castro, J. N. de Gruyter, H. Nakamura, K. Knouse, C. Qin, K. J. Clay, D. Bao, C. Li, J. T. Starr, C. Garcia-Irizarry, N. Sach, H. S. White, M. Neurock, S. D. Minteer, P. S. Baran, J. Am. Chem. Soc. 2019, 141, 6392. doi:10.1021/jacs.9b01886.















No comments yet.