

含有光氧化還原過程的雙催化反應
陽光是自然界中獨特的清潔能源,因為它廉價、無污染而且太陽無時無刻不在向外輸出光能。早在1912年,化學家Giacomo Ciamician就提出了僅使用太陽光就可以完成一個有機化學反應的設想。[1]然而,大多數有機化合物並不吸收可見光,這導致很多光化學反應都是由紫外光完成的。[2]直到2008年,普林斯頓大學的D. W. C. MacMillan以及威斯康辛大學的T. P. Yoon分別發現某些光敏劑在吸收可見光后形成相應的激發態物種通過單電子轉移 (SET) 過程活化有機物。這種新的方法在近五年得到了很大的發展。[3]
美中不足的是大多數利用可見光氧化還原催化的反應後續過程都類似於自由基反應,這也就決定了這類反應只能在“產生自由基”與尋找“自由基接受體”這兩方面做文章,極大的限制了反應底物的選擇。2011年致力於Pd催化研究的Sanford教授,利用中心金屬可以與自由基結合形成高一個氧化態的金屬配合物這一特點,完成了photoredox/ palladium共同催化的芳基化反應。這也是首例可見光-過渡金屬雙催化反應的報道。[4]光催化劑與金屬催化劑在同一個轉化中串聯運轉、各取所長,使兩種催化劑獨自不能實現的反應得到了實現,提供了化合物合成的全新的方法。此後,光催化劑 (PC) 與Au[5]、Cu[6]、Ru[7]、Co[8]等過渡金屬雙催化反應均有報導。
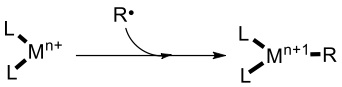
Figure 1 自由基與過渡金屬結合
光氧化還原與鎳催化劑通過單電子轉移實現2°烷基交叉偶聯
一項近期的統計表明,在藥物化學家完成的C-C鍵生成反應中,超過60%的反應可以歸類于過渡金屬催化的交叉偶聯。[9]這說明了偶聯反應對現代化學的發展極其重要,某些技術性的難題也亟待解決。目前,C(sp2)-C(sp2) 鍵偶聯的研究最為廣泛透徹。然而,C(sp2)-C(sp3)鍵的構建仍然是具有挑戰性的,很多方法並不能普遍的推廣。
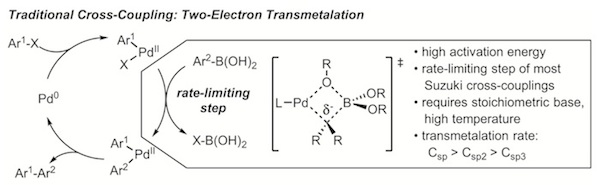
Figure 2 Pd(0) 催化C-C键偶联机理 (摘自Science, 2014, 345, 433.)
以Pd(0)為代表的交叉偶聯反應大多數是基於雙電子轉移過程:氧化加成、轉金屬、還原消除。當烷基試劑作為親核體時,轉金屬化就成為了反應的決速步。為了加快轉金屬化過程,Grignard試劑參與的Kumada-Corriu反應以及有機鋅試劑參與的Negishi反應均有所研究[10]。然而這類金屬有機試劑並不穩定,它們對空氣與潮濕敏感,操作比較困難。由於該試劑有強親核性及鹼性,此類反應官能團兼容性低,反應副產物多,整體的反應效果不甚理想。烷基硼酸或烷基氟硼酸鹽也是一類烷基親核試劑。由於有機硼試劑性能溫和且絕大多數對空氣穩定易於存放,製備硼試劑的方法較烷基金屬試劑更多,因此Suzuki-Miyaura反應備受科研工作者青睞。可是C(sp3)-硼試劑轉金屬的方法的研究還僅僅停留在在初級階段。[11]由於其親核性較弱,為提高反應產率,經常使用2-4個當量的鹼以及很高的反應溫度(~150℃),甚至是等當量的銀鹽或銅鹽。[12]於此同時,Pd作為這類反應的催化劑也有一定的局限性,即:(1)需要使用高溫條件及過量的鹼,使反應總體效益降低;(2)儘管多種Pd催化劑、配體、條件篩選等工作已經嘗試,但是諸如2-甲基烷基硼試劑這樣的高取代的脂肪基無法得到相應的偶聯化合物。
隨著可見光-金屬雙催化領域的發展,Molander教授團隊希望通過新的單電子轉移(SET)過程實現C(sp2)-C(sp3)鍵的構築。實現這個想法的一個挑戰在於,通過光氧化形成自由基,所形成的自由基需要被中心金屬俘獲使金屬被氧化,這樣才能進行接下來的光還原。Akita組早前的工作證明了在光照下以Ir[dFCF3ppy]2(bpy)PF6為催化劑可以使2°烷基氟硼酸鉀產生相應的烷基自由基。[13]金屬催化劑,從與芳基鹵反應性強且具有單電子轉移能力的Ni鹽開始研究。經優化后得到如下條件。
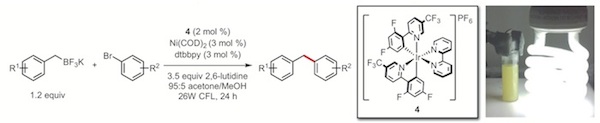
Figure 3 Molander課題組的最優條件 (摘自Science, 2014, 345, 433.)
其反應機理如下圖所示:
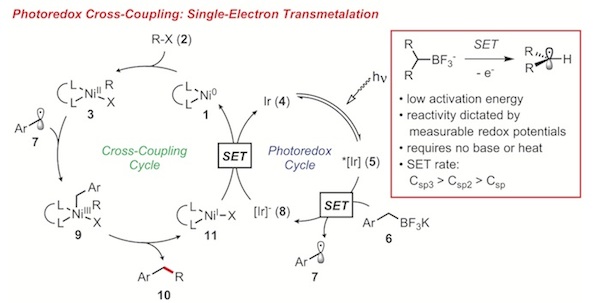
Figure 4 可見光-Ni催化循環圖 (摘自Science, 2014, 345, 433.)
光催化與Ni催化協同進行。光氧化過程產生了2°烷基自由基,金屬試劑捕獲后發生還原消除,再通多對Ni(I)的光還原使得金屬催化劑再生。
有趣的是,在Molander小組發現的同時,普林斯頓大學MacMillan教授課題組也發現了類似的反應。[14]這兩篇背靠背的工作,發表于同一期的Science雜誌中。MacMillan組使用烷基羧酸作為烷基源,在研究過程中他們發現bench-stable的NiCl2·glyme的效果比Ni(COD)2更好。其催化循環機理類似Molander的工作,但他們推測Ni(0)是光催化劑與Ni(II)發生雙電子轉移形成的,過量的氨基酸還原了光催化劑,其機理如下:
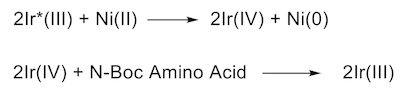
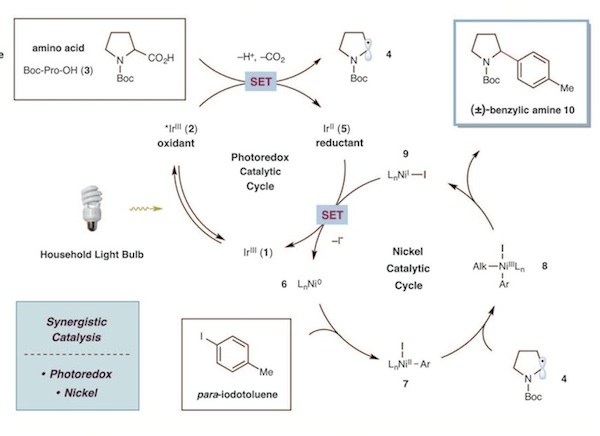
Figure 5 MacMillan課題組推測反應機理 (摘自Science, 2014, 345, 437.)
這兩個類似的反應,僅需要在惰性氛圍下,使用家用燈泡在室溫下照射,加入1%-2%光催化劑與3%-10%的鎳催化劑,通過單電子轉移過程巧妙的迴避了傳統方法裡面烷基親核試劑轉金屬化慢的問題,避免了高溫以及更加昂貴的Pd催化劑的使用。Molander小組對此反應做了一個競爭實驗,將等量的芳基硼試劑與烷基硼試劑投入反應。結果高產率得到烷基硼試劑的偶聯產物,未得到相應的芳基產物。這是由於烷基硼試劑單電子轉移速率快的原因。在另一個機理實驗中,外消旋化的烷基硼試劑在不對稱配體的調控下,反應產物得到了立體富集,而Pd催化通常會立體保持。因此,這種方法可以很好地與之前已開發的催化方法互補。
此後,Molander小組通過理論計算驗證推測的機理。[15] 通過計算,他們發現Ni催化劑可能有兩種反應途徑。除了如上所述的過程,還有可能Ni(0)先捕獲自由基,接著氧化加成,再還原消除,最後催化劑再生。
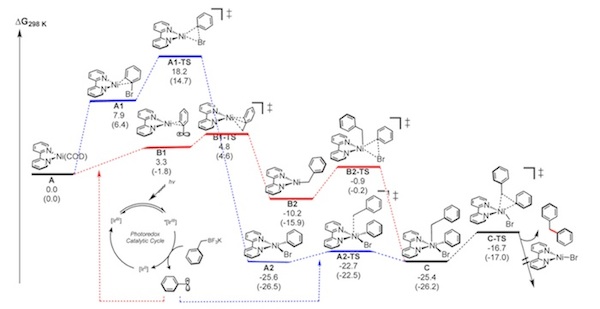
Figure 6 以2,2′-連吡啶為配體進行理論計算結果及可能的反應途徑 (J. Am. Chem. Soc. 2015, 137, 4896.)
在進一步摸清可見光氧化還原-Ni雙催化體系的機理后,Molander課題組將其在多種類型2°烷基硼試劑進行了一系列的拓展,獲得了較好的產率,證明了此方法具有可觀的普適性。[16]-[19]
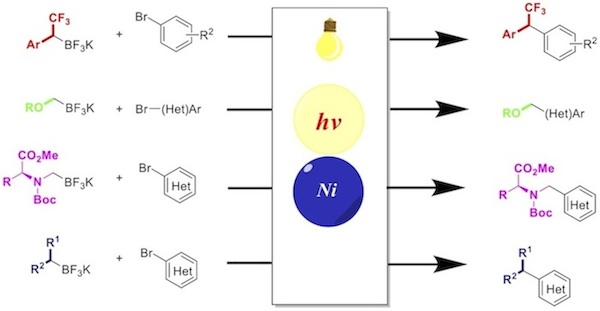
Figure 7 Molander課題組對雙催化烷基化反應的拓展
Molander教授很興奮的表示,這種方法極大的簡化了反應的操作也極大的推動了2°烷基交叉偶聯反應的發展。但唯一的缺點是,由於烷基自由基的產生,該方法可能使原本具有立體專一性的烷基外消旋化,如何通過配體調控解決這一問題將是該課題組以後工作的重點。
相关文献
- T. P. Yoon, M. A. Ischay, J. Du Nature Chem., 2010, 2, 527. DOI: 10.1038/nchem.687
- N. Hoffmann, Chem. Rev. 2008, 108, 1052. DOI: 10.1021/cr0680336
- C. K. Prier, D. A. Rankic, D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322. DOI: 10.1021/cr300503r
- D. Kalyani, K. B. McMurtrey, S. R. Neufeldt, M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566. DOI: 10.1021/ja208068w
- A. Tlahuext-Aca, M. N. Hopkinson, B. Sahoo, F. Glorius, Chem. Sci., 2016, 7, 89. DOI: 10.1039/C5SC02583D
- Y. Ye, M. S. Sanford J. Am. Chem. Soc., 2012, 134, 9034. DOI: 10.1021/ja301553c
- D. C. Fabry, M. A. Ronge, J. Zoller, M. Rueping, Angew. Chem. Int. Ed., 2015, 54, 2801. DOI: 10.1002/anie.201408891
- G. T. Zhang, C. Liu, H. Yi, Q. Y. Meng, C. L. Bian, H. Chen, J. X. Jian, L. Z. Wu, A. Lei, J. Am. Chem. Soc., 2015, 137, 9273. DOI: 10.1021/jacs.5b05665
- S. D. Roughley, A. M. Jordan J. Med. Chem., 2011, 54, 3451. DOI: 10.1021/jm200187y
- E. Jahn, U. Jahn, Angew. Chem. Int. Ed., 2014, 53, 13326. DOI: 10.1002/anie.201408748
- J. C. Tellis, D. N. Primer, G. A. Molander, Science, 2014, 345, 433. DOI: 10.1126/science.1253647
- (a) D. Imao, B. W. Glasspoole, V. S. Laberge, C. M. Crudden, J. Am. Chem. Soc., 2009, 131, 5024; DOI: 10.1021/ja8094075 (b) J. Z. Deng, D. V. Paone, A. T. Ginnetti, H. Kurihara, S. D. Dreher, S. A. Weissman, S. R. Stauffer, C. S. Burgey, Org. Lett., 2009, 11, 345. DOI: 10.1021/ol802556f
- (a) Y. Yasu, T. Koike M. Akita* Adv. Syn. Cat., 2012, 354, 3414; DOI: 10.1002/adsc.201200588 (b) K. Miyazawa, Y. Yasu, T.Koike, M. Akita, Chem. Comm., 2013, 49, 7249. DOI: 10.1039/C3CC42695E
- Z. W. Zuo, D. T. Ahneman, L. L. Chu, J. A. Terrett, A. G. Doyle, D. W. C. MacMillan, Science, 2014, 345, 437. DOI: 10.1126/science.1255525
- O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander, M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896. DOI: 10.1021/ja513079r
- D. Ryu, D. N. Primer, J. C. Tellis, G. A. Molander, Chem. Eur. J., 2016, 22, 120. DOI: 10.1002/chem.201504079
- I. Karakaya, D. N. Primer, G. A. Molander, Org. Lett., 2015, 17, 3294. DOI: 10.1021/acs.orglett.5b01463
- M. E. Khatib, R. Augusto, M. Serafim, G. A. Molander, Angew. Chem. Int. Ed., 2016, 55, 254. DOI: 10.1002/anie.201506147
- D. N. Primer, I. Karakaya, J. C. Tellis, G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195. DOI: 10.1021/ja512946e
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
















No comments yet.