
作者:石油醚
导读:
中国医学科学院/北京协和医学院医药生物技术研究所付海根课题组采用酶催化动态动力学拆分策略,利用亚胺还原酶对底物的泛杂性和定向进化工具,实现了轴手性联芳基化合物的不对称合成,拓展了酶催化可及空间。相关成果在Angew. Chem. Int. Ed. 发表。
“Atroposelective Synthesis of Axial Biaryls by Dynamic Kinetic Resolution Using Engineered Imine Reductases.
Xinyue Hao, Zhuangfei Tian, Zhouchang Yao, Tienan Zang, Shucheng Song, Liang Lin, Tianzhang Qiao, Ling Huang, Haigen Fu*
Angew. Chem. Int. Ed. 2024, e202410112. Doi: 10.1002/anie.202410112”
正文:
轴手性联芳基化合物是许多天然产物、生物活性分子、手性配体和催化的核心骨架,如抗生素万古霉素,抗白血病化合物Steganacin,手性配体BINOL和有机催化剂CPAs(图1)。现已发展了诸多构建轴手性联芳基化合物的不对称化学催化方法,主要包括传统交叉偶联、芳基C-H活化、氧化交叉偶联、原位构环、中心到轴向手性转移等策略。然而,这些方法通常需要使过渡金属催化剂、复杂手性配体或助剂、预先安装导向基团或使用有机溶剂等。鉴于轴手性联芳基化合物的广泛应用,亟待进一步发展高效且可持续的催化方法来合成轴手性联芳基化合物。
酶催化具有高率、高选择性和易于进化的特点。虽然酶催化已被广泛认可并用于构建许多药物和精细化学品的手性中心,但其用于构建手性轴的潜力尚未得到充分挖掘。目前制备轴手性联苯的生物催化策略主要包括:(1)两个芳基片段的直接偶联;(2)对前手性或外消旋联芳基前体的后期转化(图1B)。然而,轴手性联苯的生物催化不对称合成通常局限于少数几种酶类(如酯水解酶、酮还原酶、卤化酶等),限制了反应类型和产物的多样性。因此,开发全新高效、高选择性的生物催化方法来合成高价值的轴手性联苯化合物具有重要意义。
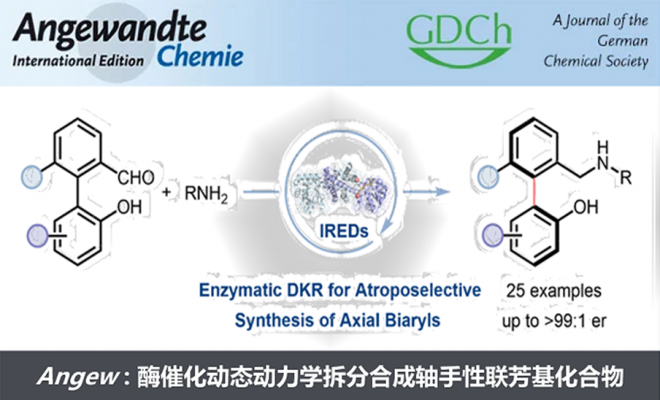
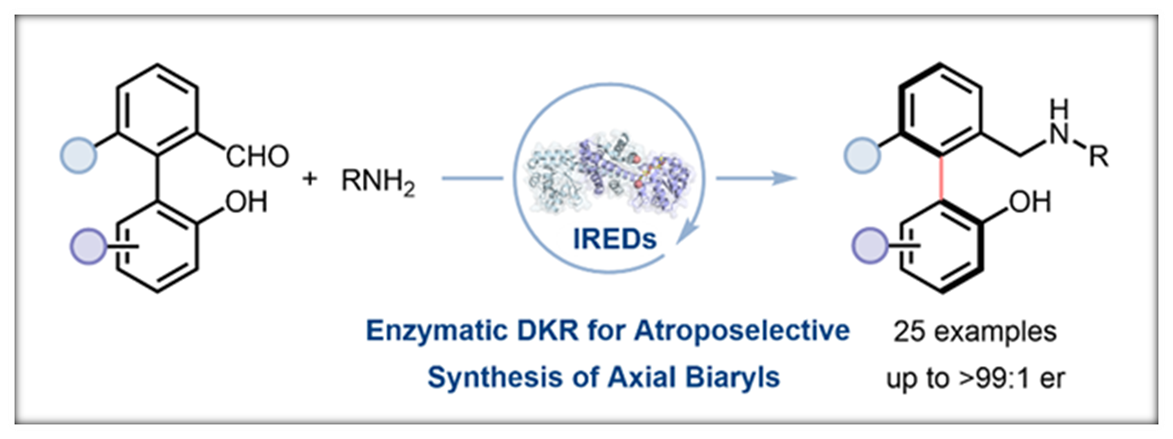
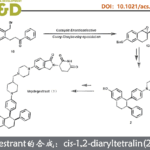
在本项研究中,中国医学科学院/北京协和医学院医药生物技术研究所付海根课题组报道了一种新的基于动态动力学拆分(DKR)亚胺还原酶(IRED)催化的不对称合成轴手性联芳基化合物方法(图1C)。受桥联轴手性联芳基化合物构型不稳定易于发生旋转消旋化的启发,研究者设想:消旋联芳基羟基醛原料1与胺原位生成联芳基亚胺(外消旋体3和5),随后通过可逆形成构型不稳定的N,O-缩醛桥联化合物4,从而使外消旋体3和5发生动态相互转化,最后利用IRED对联芳基亚胺5进行立体选择性还原拆分,得到所需的轴手性联芳基产物2(图1C)。
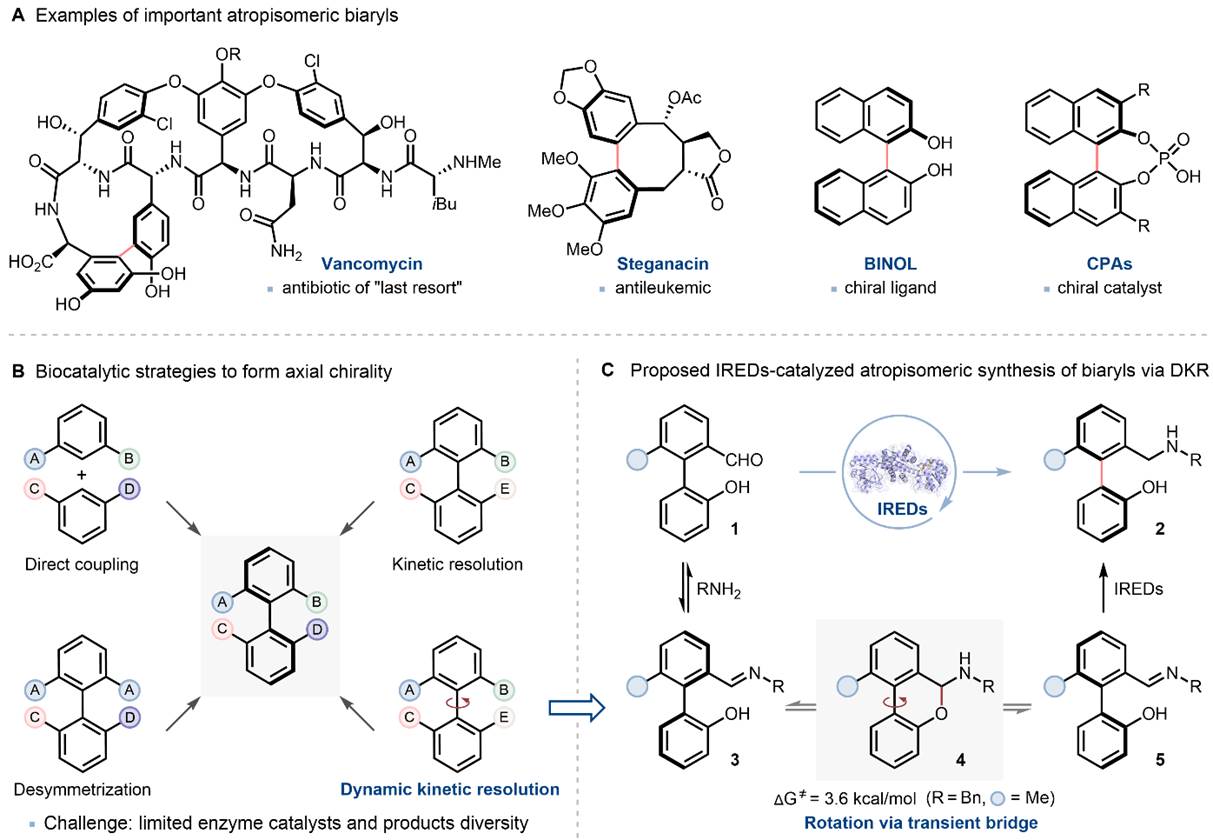
图 1. 生物催化合成轴手性联芳基化合物。图片来源:Angew. Chem. Int. Ed.
首先,作者以外消旋联芳基羟基醛6a和苄胺7a为模板底物,筛选了一系列野生型IRED和还原氨基酶(RedAm),发现大多酶都可以催化反应得到少量的目标产物联芳基氨基醇8。其中,一个来自Streptomyces sp. GF3546的S-IRED-Ss能够以35%的产率和94:6的er值得到(S)-型目标产物8。通过将S-IRED-Ss蛋白浓度从0.2 mol%增加到1 mol%,可将联芳基8的产率提高至76%(超过动力学拆分50%的理论最大产率),且立体选择性不降低,验证这是一生物催化DKR过程的猜想(图2A)。前人对S-IRED-Ss蛋白非催化位点改造获得了一个高度进化的9突变体(S-IRED-Ss-M9),该突变体具有更高的热稳定性和耐有机溶剂性质。S-IRED-Ss-M9突变体也能够催化联芳基产物8的不对称合成,当酶量为0.2 mol%时,产率为43%,er值为93:7,这为蛋白定向进提供了一个良好的起点(图2A)。随后,作者采用迭代饱和突变的策略对S-IRED-Ss-M9进行改造,经过两轮进化,找到基于M9的双突变体S-IRED-Ss-M11(M236T/P122N)可高效、高立体选择性地催化反应得到8(97%,97:3 er),活力相对于野生型提高了8.6倍,且该反应可以用粗酶的冻干粉放大到1.0 mmol规模(77%,97:3 er,234 mg)(图2B)。
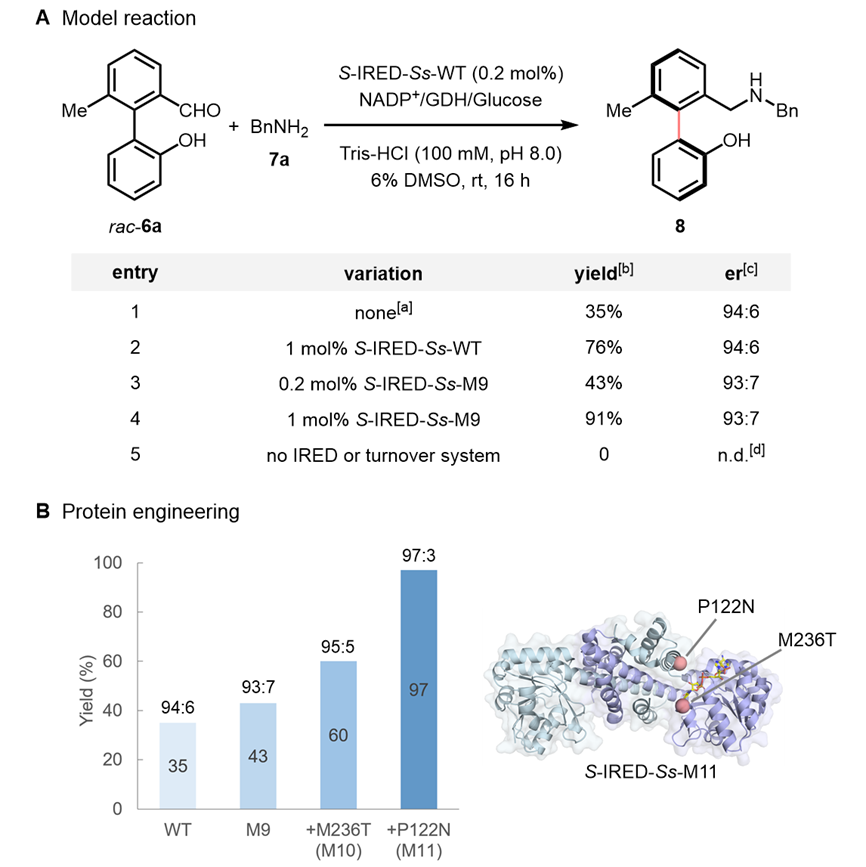
图 2. 模板反应优化。图片来源:Angew. Chem. Int. Ed.
随后,作者对S-IRED-Ss-M11的底物范围进行了探索。对于反应的胺谱,多种芳基取代的苄胺均可很好地与底物6a发生还原反应。如苯环的间位和对位可兼容带有不同电性的取代基,以59–98%的产率以及优异的对映选择性(>93:7 er)得到轴手性联芳基氨基醇(10-17)。此外,S-IRED-Ss-M11还可兼容吡啶杂环、烯丙胺、炔丙胺等一级胺,得到相应的产物(18-20),具有良好的收率(57–94%)和高对映选择性(最高99:1 er),但是对于二级胺或芳香胺则不兼容(图3)。该酶反应的联芳酚醛的底物范围也很广泛,S-IRED-Ss-M11可接受在醛芳环上各类取代的联芳基酚醛为底物,得到相应的轴手性联芳基氨基醇产物21-23(产率30–88%,最高92:8 er),但在该醛芳环上进行二甲基或萘基取代则耐受性较差。对于酚环底物的范围而言,在不同位置添加各类取代基的底物均可被酶接受,产率为35–88%,对映选择性最高可达99:1 er。遗憾的是,酶催化构建双轴手性产物未能成功。
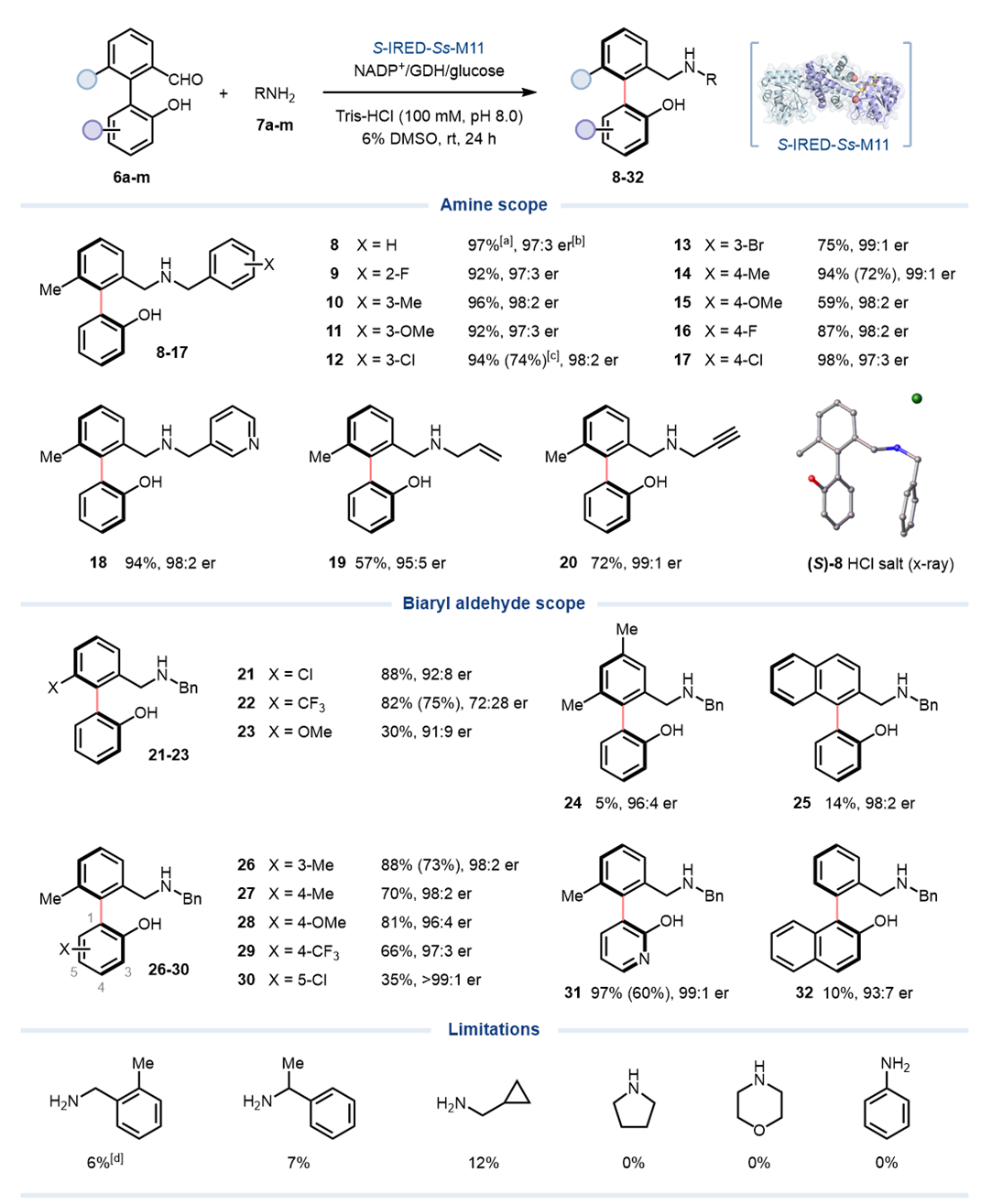
图 3. 底物范围。图片来源:Angew. Chem. Int. Ed.
最后,作者采用分子动力学(MD)模拟对S-IRED-Ss-M11突变体的活性和立体选择性提高进行了解释。MD模拟显示,野生型S-IRED-Ss更倾向于与亚胺中间体(S)-33发生反应,因为(S)-33亚胺碳C8到辅酶NADPH烟酰胺环C4的距离比相应的(R)-33亚胺碳C8到辅酶NADPH C4的距离要短,结合能也更低(4.5 Å,-26.4 kcal/mol 对比6.1 Å, -20.7 kcal/mol)。对S-IRED-Ss-M11突变体,虽然活性位点未观察到明显的构象变化,但与野生型酶相比,相应辅酶NADPH C4原子到(S)-33 C8原子之间的距离和结合能(3.6 Å,-33.3 kcal/mol)均更有利于氢负转移的发生。另外,对于M11突变体,潜在质子供体Y169的酚氧原子与底物(S)-33的氮原子之间的距离比相应的野生型酶更短,这也有利于亚胺还原的最后质子化步骤。
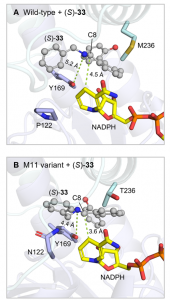
图 4. 分子动力学模拟。图片来源:Angew. Chem. Int. Ed.
总结,本文采用酶催化动态动力学拆分策略,利用亚胺还原酶对底物的泛杂性和定向进化工具,实现了轴手性联芳基化合物的不对称合成,拓展了酶催化可及空间。相关成果在Angew. Chem. Int. Ed.上发表,中国医学科学院/北京协和医学院医药生物技术研究所研究生郝新月、田壮飞、姚周畅为本文的共同一作,黄玲博士及付海根研究员为共同通讯作者,中科院上海有机所林亮研究员为本研究提供了帮助。
(感谢付海根研究员课题组对Chem-Station的支持,感谢文章作者郝新月提供稿件)
招生信息:付海根,现任中国医学科学院/北京协和医学院医药生物技术研究所研究员,目前以第一或通讯作者在Nature, Nature Catalysis, JACS, Angew等高水平期刊上发表论文十余篇,入选2022年国家海外青年人才计划。课题组的主要研究方向为生物催化及药物化学。欢迎感兴趣的同学报考2025年入学的硕士(保研)和博士研究生,请发送相关材料到付老师邮箱fuhaigen@hotmail.com。相关链接:付海根 (imb.com.cn)。















No comments yet.