
本文作者:杉杉
导读
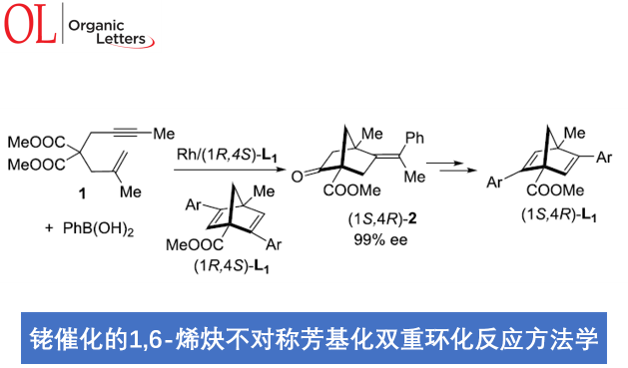
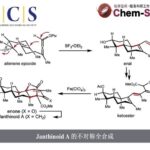
近日,台湾清华大学的T. Hayashi (林 民生, Hayashi Tamio)与内蒙古大学的明佳林课题组合作报道一种铑催化的1,6-烯炔化合物 (1)参与的不对称芳基化双重环化 (asymmetric arylative biscyclization)反应方法学,进而成功完成一系列具有双环[2.2.1]庚二烯骨架以及桥头碳原子中存在甲基与酯基取代的新型手性二烯配体 (1R,4S)-L1的构建。之后,作者发现,手性二烯配体 (1R,4S)-L1能够进一步应用于铑催化剂促进的1,6-烯炔化合物1的不对称双重环化反应,并获得99% ee的双环产物 (1S,4R)-2。同时,研究发现,(1S,4R)-2能够进一步转化为手性二烯配体 (1S, 4R)-L1。
Asymmetric Synthesis of Chiral Bicyclo[2.2.1]hepta-2,5-diene Ligands through Rhodium-Catalyzed Asymmetric Arylative Biscyclization of a 1,6-Enyne
C. Sun, H. Meng, C. Chen, H. Wei, J. Ming, T. Hayashi, Org. Lett. 2021, 23, 6311. doi: 10.1021/acs.orglett.1c02088.
正文
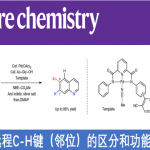
手性二烯化合物是过渡金属催化的不对称反应方法学研究中的重要配体。其中,具有双环[2.2.1]庚二烯 (nbd)骨架的手性二烯配体已经广泛应用于一系列涉及铑催化剂促进的不对称芳基化反应的相关研究,并实现最佳的对映选择性控制 (Figure 1a)。2003年,通过钯催化剂促进的对映纯降冰片烷二醇 (norbornanediol)参与的不对称硅氢化反应方法学,首次实现具有C2-对称性的手性配体 (R,R)-Bn-nbd的合成[1]。同时,Corey课题组报道采用不对称Diels-Alder反应策略实现单取代nbd配体的制备[2]-[3]。之后,Wu课题组报道采用(-)-乙酸冰片酯 ((-)-bornyl acetate)作为起始原料,进而成功实现完成一系列具有假C2-对称性的nbd配体的构建[4]。此外,本课题组前期已经报道一种采用铑催化剂参与的1,6-烯炔与芳基硼酸之间的芳基化环化反应策略[5]-[6],并以较高的反应收率获得相应的双环[2.2.1]庚烷衍生物 (Figure 1b)。之后,Murakami (村上正浩, Murakami Masahiro)课题组选择手性的Rh/(R)-binap作为催化剂,成功设计出一种具有高度对映选择性的芳基化环化反应方法学[7]。受到上述研究报道的启发,这里,作者成功开发出如下三种较为重要的合成转化策略:(1) 采用手性二烯配体提高不对称芳基化环化反应过程中的化学与对映选择性 (2) 将对映富集的双环[2.2.1]庚烷产物转化为新型的手性nbd配体 (3) 将手性nbd配体进一步应用于铑催化的不对称芳基化反应方法学的相关研究 (Figure 1c)。

其中,Murakami团队[7]报道一种铑催化的1,6-烯炔衍生物1与苯硼酸之间的环化反应,进而获得60%收率与89% ee的手性双环庚-2-酮产物2。然而,由于上述反应过程中存在通过Ph-Rh中间体与1中三键的加成过程,并进一步形成副产物四氢萘酮衍生物3,进而使反应收率显著降低。为解决上述环化过程中目标产物收率较低的问题,这里,作者采用1,6-烯炔衍生物1与PhB(OH)2或PhZnCl之间的环化过程作为模型反应,进行了相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用[RhCl(L2a)]2作为催化剂,PhZnCl作为芳基化试剂,在THF反应溶剂以及反应温度为50 oC的条件下进行反应,最终获得89%反应收率与98% ee的环化产物 (1R,4S)-2。并且,反应过程中无副产物3的形成。

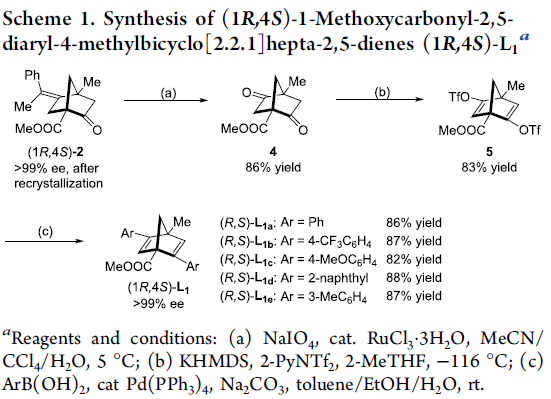
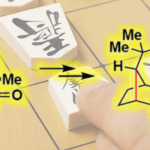
之后,作者发现,通过在正己烷溶剂中进行的重结晶操作,能够进一步使环化产物(1R,4S)-2的对应纯度提高至>99% ee。值得注意的是,对映纯的烯酮产物 (1R,4S)-2能够通过后续的三步反应过程,进一步转化为双环[2.2.1]庚二烯配体L1 (Scheme 1)。首先,通过钌催化对烯基氧化过程,获得86%反应收率的二酮4。之后,将二酮4进一步通过KHMDS与2-PyNTf2处理,获得83%收率的双三氟甲磺酸酯砌块5。接下来,选择钯催化剂促进的三氟甲磺酸酯砌块5与苯硼酸之间的交叉偶联过程,最终获得较高反应收率并> 99% ee的手性二烯配体(1R,4S)-L1a-1e。值得注意的是,这里合成出的手性二烯配体 (1R,4S)-L1 尽管具有与Ph-nbd以及Wu课题组发展的L3配体相类似的骨架结构,然而,不同之处则在于(1R,4S)-L1配体的桥头碳原子中分别具有甲基与酯基的取代,并且在空气或CDCl3中能够稳定存在数日。
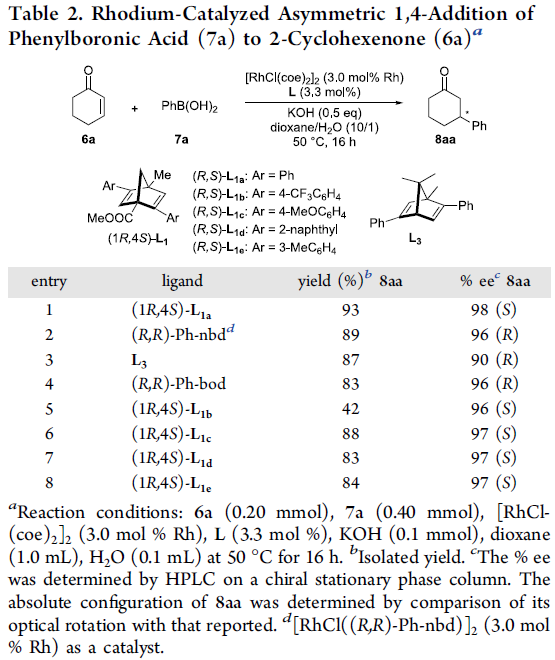
接下来,作者将这一全新的手性二烯配体 (1R,4S)-L1应用于铑催化剂存在下,PhB(OH)2与2-环己烯酮 (6a)之间的不对称共轭加成反应的研究,并对反应过程的对映选择性进行考察 (Table 2)。研究表明,采用[RhCl(coe)2]2作为催化剂,(1R,4S)-L1a作为手性配体,在二氧六环/H2O的混合溶剂中,反应温度为50 oC,反应时间为16 h,最终获得93%收率与98% ee的加成产物S–8aa。同时,作者发现,采用Ph-nbd、L3以及Ph-bod作为配体时,则获得相反构型的加成产物R–8aa (90-96% ee)。
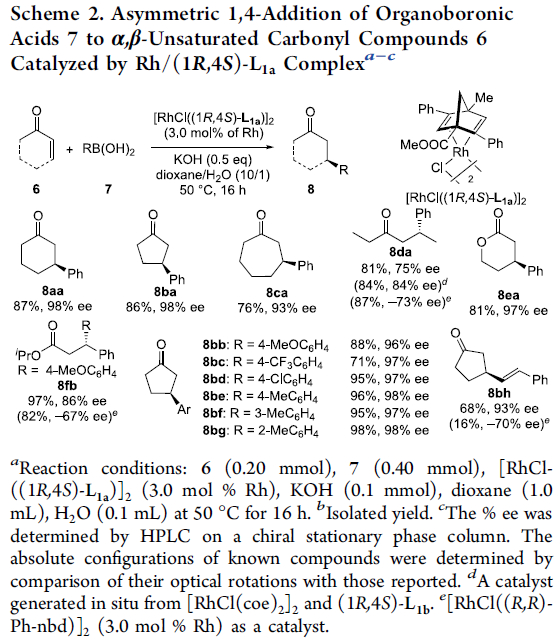
为进一步研究上述不对称共轭加成过程中的底物应用范围 (Scheme 2),作者分别选择具有氯桥配位的Rh催化剂二聚体(chloro-bridge dimer) [RhCl((1R,4S)-L1a)]2 (Table 2)以及 [RhCl(coe)2]2配合物与手性配体 (1R,4S)-L1a之间原位形成的手性钌配合物作为催化剂,进行上述不对称共轭加成过程的研究。该小组观察到,除2-环己烯酮 (6a)之外,2-环戊烯酮 (6b)、2-环庚烯酮 (6c)以及环烯酸酯底物 (6e),均能够获得具有较高反应收率与高度对映选择性 (93-98 % ee)的目标产物8ba、8ca以及8ea。而对于直链烯酮6d与烯酸酯底物6f,则观察到对映选择性的显著降低。接下来,作者对芳基硼酸底物的应用范围进行深入研究。实验观察到,一系列芳基中不同位置带有各类不同取代基团的苯硼酸底物,均能够顺利地参与上述的不对称共轭加成过程,并获得相应的氢芳基化产物8bb–8bg (71-98% 反应收率, 96-98% ee)。同时,该小组发现,采用反式-2-苯基乙烯基硼酸(7h)作为反应底物时,同样能够将烯基顺利引入至共轭烯酮底物中,并以优良的对映选择性 (93% ee)获得相应的手性产物8bh。值得注意的是,在相同的反应条件下,采用[RhCl((R,R)-Ph-nbd)]2作为催化剂,则观察到较低的对映选择性控制,例如产物8da、8fb以及8bh。同时,作者通过对[RhCl((1R,4S)-L1a)]2与[RhCl((R,R)-Ph-nbd)]2之间X-射线单晶结构的比较,进而对上述反应过程中对映选择性的控制因素进行深入研究 (详见Supporting Information)。
此外,作者选择(1R,4S)-L1作为手性配体,对1,6-烯炔衍生物参与的不对称芳基化环化反应方法学进行研究,进而进一步阐明手性二烯配体的self-replication过程 (Table 3)。实验结果表明,在采用(1R,4S)-L1e作为手性配体时,1,6-烯炔衍生物1与PhB(OH)2之间的环化过程,能够获得83%收率与99% ee的手性产物 (1S,4R)-2。并且,手性产物 (1S,4R)-2同样能够继续通过Scheme 1中的三步反应过程,转化为对映纯的二烯配体 (1S,4R)-L1a。通过上述的反应过程,进而成功实现互为对映体的两种手性二烯配体之间的self-replication。因此,选择手性二烯配体的其中一种对映体,经历上述的芳基化环化以及后续的转化步骤之后,能够最终获得这一手性二烯配体的对映体。同时,作者观察到,选择L3作为手性配体时,能够获得60%收率与87% ee的具有相反构型的对映纯产物 (1R,4S)-2。

总结
台湾清华大学T. Hayashi与内蒙古大学明佳林课题组共同报道一种新型的具有双环[2.2.1]庚二烯骨架以及桥头碳原子中具有甲基与酯基取代基团的手性二烯配体 (1R,4S)-L1的合成设计策略。并且,作者发现,这一新型的手性二烯配体在铑催化的芳基或烯基硼酸与α,β-不饱和羰基化合物之间的不对称共轭加成过程中,能够表现出优良的催化活性与高度的对映选择性控制。此外,这一全新的手性二烯配体 (1R,4S)-L1同样能够进一步应用于铑催化的1,6-烯炔衍生物1的不对称芳基化双重环化反应的研究,并获得99% ee的双环产物 (1S, 4R)-2。而且,作者进一步发现,双环产物(1S, 4R)-2能够进一步作为合成手性二烯配体(1R, 4S)-L1的重要前体。
参考文献
[1] (a) T. Hayashi, K. Ueyama, N. Tokunaga, K. Yoshida, J. Am. Chem. Soc. 2003, 125, 11508. doi: 10.1021/ja037367z.(b) G. Berthon-Gelloz, T. Hayashi, J. Org. Chem. 2006, 71, 8957. doi: 10.1021/jo061385s.
[2] M. K. Brown, E. J. Corey, Org. Lett. 2010, 12, 172. doi: 10.1021/ol9025793. [3] K. Okamoto, T. Hayashi, V. H. Rawal, Org. Lett. 2008, 10, 4387. doi: 10.1021/ol801931v. [4] W. T. Wei, J. Y. Yeh, T. S. Kuo, H. L. Wu, Chem. – Eur. J. 2011, 17,11405. doi: 10.1002/chem.201102073. [5] R. Shintani, K. Okamoto, Y. Otomaru, K. Ueyama, T. Hayashi, J. Am. Chem. Soc. 2005, 127, 54. doi: 10.1021/ja044021v. [6] (a) E. Negishi, C. Copéret, S. Ma, S. Y. Liou, F. Liu, Chem. Rev. 1996, 96, 365. doi: 10.1021/cr950027e.(b) C. Aubert, O. Buisine, M. Malacria, Chem. Rev. 2002, 102, 813. doi: 10.1021/cr980054f.
[7] T. Miura, T. Sasaki, H. Nakazawa, M. Murakami, J. Am. Chem. Soc. 2005, 127, 1390. doi: 10.1021/ja043123i.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!















No comments yet.