

作者:石油醚
引言
癫痫是一种由中枢神经系统异常引起的慢性脑部疾病,表现为无诱因的癫痫发作、行为或感觉异常,有时伴随意识丧失。全球约有5000万人患病,是世界上最常见的神经系统疾病之一。γ-丁内酰胺衍生物广泛存在于自然界。(S)-Levetiracetam(1,商品名开浦兰,UCB S.A.)属于γ-丁内酰胺衍生物,是一种广谱抗癫痫药,且属于”第二代”抗癫痫药,常用于儿童和成人的附加治疗。其确切作用机制尚不明确,但已知可通过结合突触小泡蛋白SV2A,减少神经递质释放并抑制突触前Ca²⁺通道,作用方式不同于传统抗惊厥药物。
图1左乙拉西坦(开浦兰/Keppra)
左乙拉西坦(开浦兰/Keppra):前世今生
(S)-Levetiracetam(1,商品名开浦兰)是一种吡咯烷酮衍生物,化学结构与经典抗癫痫药物完全不同,属于”第二代”抗癫痫药。它最早由比利时UCB公司研发,2000年在美国上市,2007年正式进入中国,商品名为”开浦兰”。因其英文名Levetiracetam,临床上常被简称为”LEV”。目前已有多种关于该化合物的合成方法报道。合成1的方法可根据手性引入方式分为三类:手性池法、不对称合成法和拆分法。
左乙拉西坦(开浦兰/Keppra)的合成路线
1、[手性池法]
1.1 (S)-2-aminobutanamide hydrochloride (2)作为原料
1可由2经一至两步反应制得,具体取决于所用碱的种类。在KOH、TBAB催化及Na2SO4存在下,化合物2与4-氯丁酰氯在二氯甲烷(DCM)中反应,得到目标产物1(74% yield)。该反应通常经N-酰化后直接环合,无需分离中间体(Scheme 1)。然而,该路线存在若干缺陷:碱性较强无机碱会导致消旋化,且因KOH和Na2SO4过量会生产固废。有机碱可产率提升至85%,但需增加4-氯丁酰氯的用量。改用4-溴丁酰溴可采用更温和的碱,减少外消旋化。
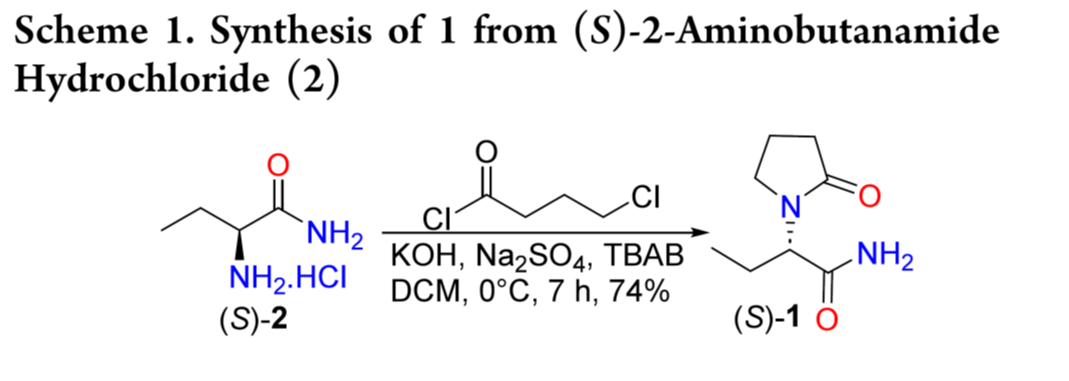
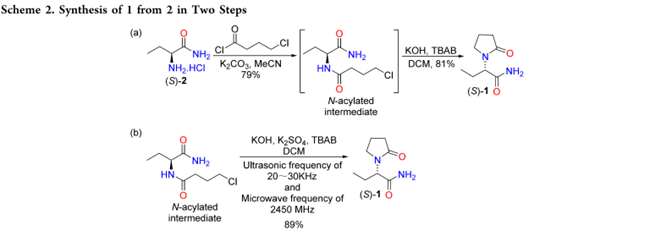
Gobert和Shen分别报道了以碳酸钾为碱,由 2 合成 1 的方法。即2在MeCN中用K2CO3分离获得 N-酰化产物,随后用KOH 和 TBAB发生环合。主要区别在于:Shen使用 TBAB,产率达 81%,高于Gobert(47% yield)。其中,第一步中加入TBAB 作为相转移催化剂,可促进碱溶入反应体系,加快反应速率,从而减少 KOH 用量,降低酰胺水解为酸的风险,提高产率(Scheme 2a)。sun等人采用 20–30 kHz 超声与 2450 MHz 微波促进环合,产率达 89%(Scheme 2b)。
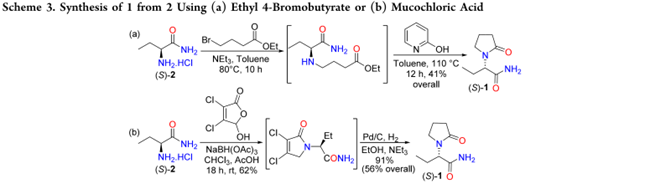
Gan等使用2和4-氯丁酸甲酯,在异丙醇中以三乙胺为碱反应36 h,得到1,但产率仅为61%。从 2 合成 1 的其他方法包括使用 4-溴丁酸乙酯和粘氯酸。在TEA存在下, 2 与 4-溴丁酸乙酯在甲苯中反应生成 N-单烷基化中间体,随后在 2-羟基吡啶作用下环合得 1(Scheme 3a)。该法需使用昂贵的 2-羟基吡啶,且反应时间长、产率低。Das Sarma 等通过粘氯酸与 2 发生还原胺化构建 1 中的 2-吡咯烷酮结构,再经脱卤和氢化得到 1(Scheme 3b)。
1.2 (S)-2-Aminobutanoic Acid (3)作为原料
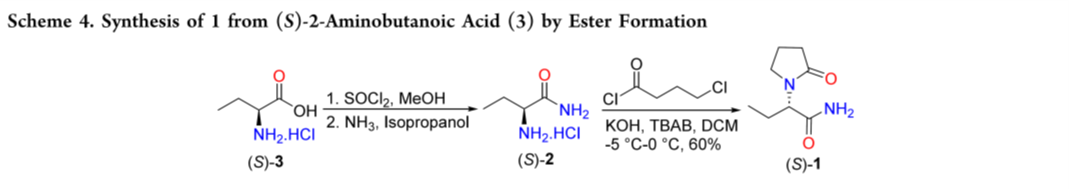
(S)-2-Aminobutanoic Acid (3)作为原料。酸转化为酰胺可通过酯化、混合酸酐法或 Boc酸酐活化后氨解实现。Acharyulu等人以3原料经酯化和氨解生成 2,随后在氢氧化钾存在下与 4-氯丁酰氯反应得到 1 (60 %)(Scheme 4)。该方法的主要缺点是产率低,并产生有害酸性废液。
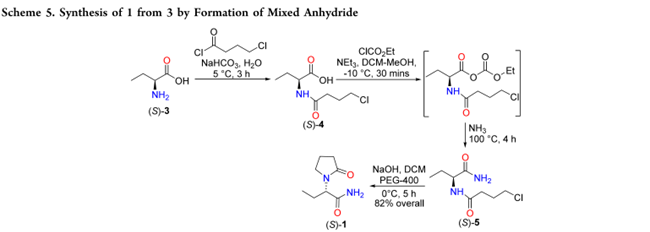
Xu等人利用3, 经混合酸酐法合成 1:酸 3 先与 4-氯丁酰氯反应得(S)-4,再与氯甲酸乙酯生成混合酸酐;该中间体经氨解得(S)-5,随后环合得到 1(82% yield,Scheme 5)。但氯甲酸乙酯剧毒,存在安全风险且增加成本。
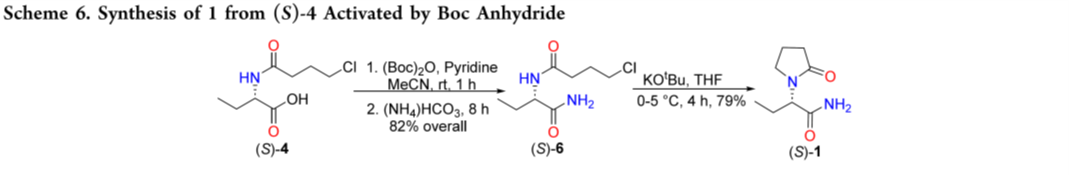
Yu等人报道由 3 合成 1 的方法(65% overall yield)。3 与 4-氯丁酰氯反应得 (S)-4;(S)-4 经 (Boc)₂O 活化后,用碳酸氢铵氨解生成 (S)-6,再在 THF 中以叔丁醇钾为碱环合得到 1(Scheme 6)。
1.3 (S)-2-Aminobutanol(7)作为原料
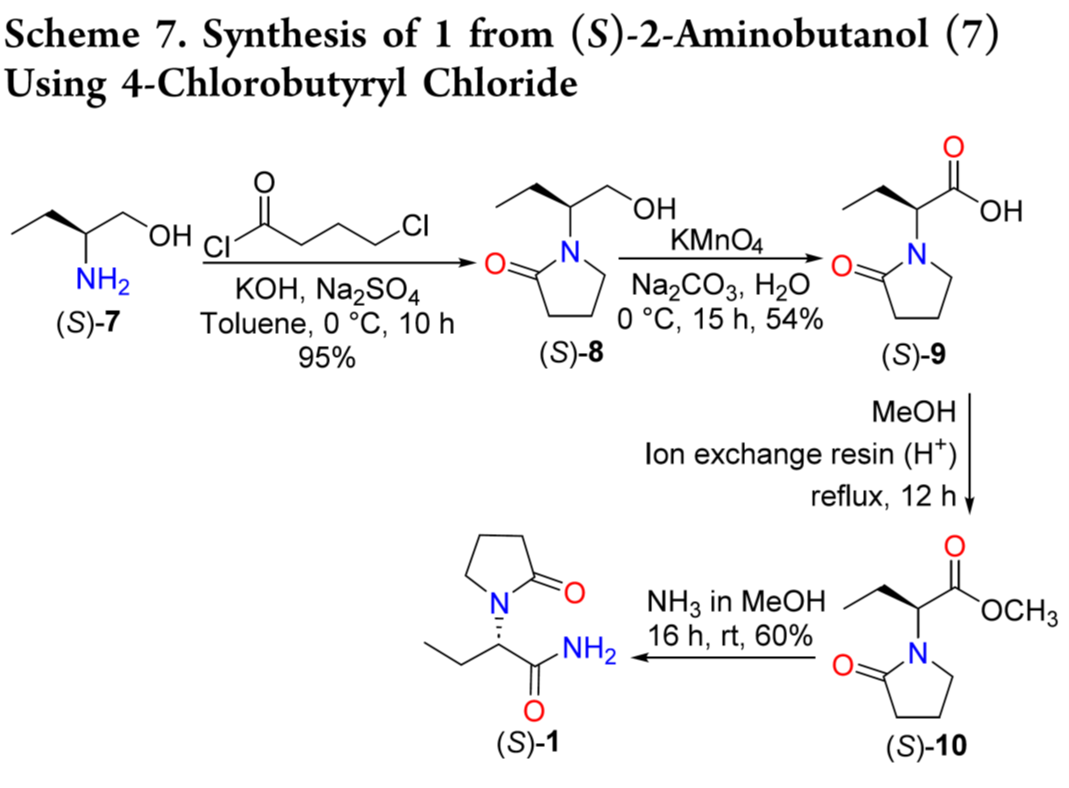
7与 4-氯丁酰氯环合得(S)-8,再经高锰酸钾氧化生成(S)-9。9经酯化后氨解,最终以 31% 总收率得到 1(Scheme 7)。该路线步骤多、产率低,且使用有毒的高锰酸钾,不利于放大生产。
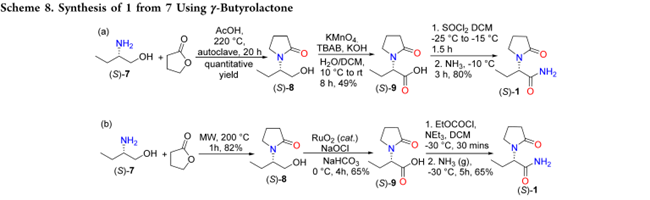
另外一方面,7 与 γ-丁内酯的混合物在高压釜中用乙酸处理或在微波反应器中于 200 °C 加热即可得到(S)-8。8再经高锰酸钾或 RuO₂/NaOCl 氧化生成酸(S)-9。(S)-9通过 SOCl₂ 制备酰氯或与烷基卤甲酸酯形成混合酸酐,随后氨解,分别以 39% 和 35% 的总收率得到 1(Sceheme 8a,b)。该路线步骤较多,且使用刺激性试剂 RuO₂ 和腐蚀性液体 SOCl₂,不利于实际应用。
1.4 L-Threonine作为原料
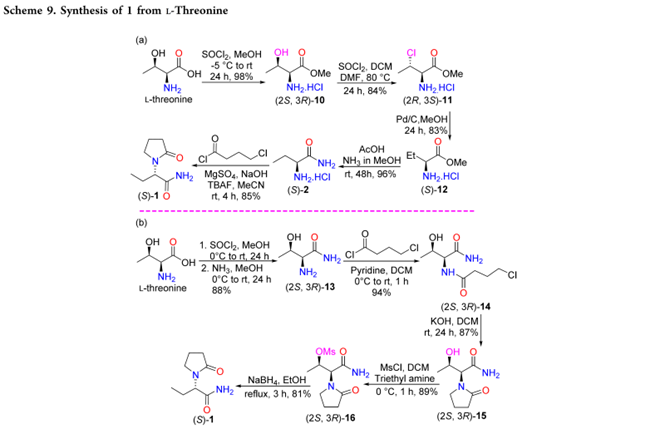
Shen和He以L-Threonine作为原料合成1。其中, L-Threonine中羧基经酯化、取代和Pd-C消除氯得到(S)-12;12经氨解为2,再与4-氯丁酰氯反应,以56%的总收率得到1(Scheme 9a)。另外一种,L-Threonine中羧基经酯化和氨解得到酰胺,在与4-氯丁酰氯反应生成3-羟基左乙拉西坦((2S,3R)-15);其羟基经甲磺酰化后,用NaBH4还原消除甲磺基,以59%的总收率得到1(Scheme 9b)。该路线中使用的亚硫酰氯和吡啶可能不适用于工艺放大。
1.5 L-Methioninee作为原料
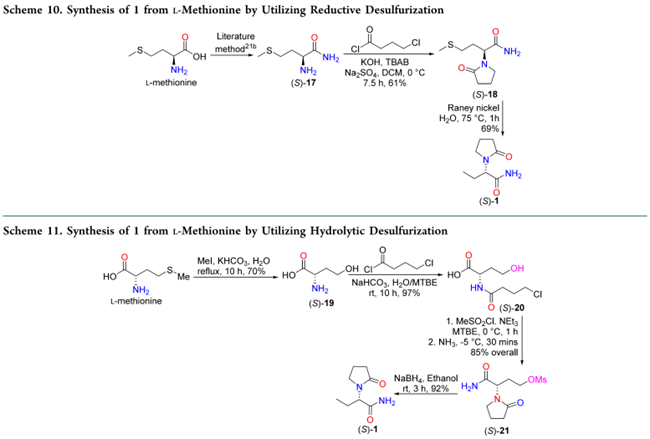
Cossement等人以文献方法制得(S)-2-氨基-4-(甲硫基)丁酰胺((S)-17),再与4-氯丁酰氯反应生成4-甲硫基列维他西坦((S)-18),随后经雷尼镍T-1脱硫,以42%的总收率得到1(Scheme 10)。Shen等人采用另一路线:将L-甲硫氨酸的硫甲基氧化为羟基,得(S)-19;其氨基与4-氯丁酰氯发生酰化生成(S)-20;再将羟基甲磺酰化,继而羧基经酰胺化并环合,形成4-O-甲磺酰左乙拉西坦((S)-21);最后用NaBH4还原消除甲磺酰基,以53%的总收率获得1(Scheme 11)。
2、[不对称合成法]
2.1 不对称氰化
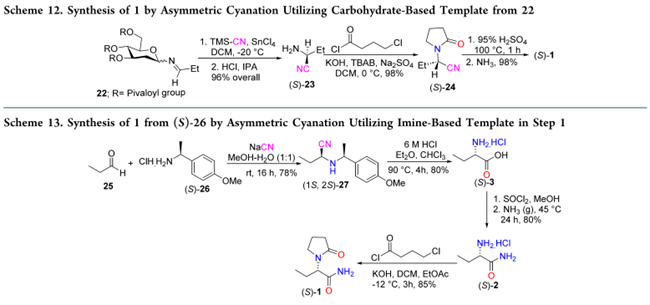
Zhang采用N-(3,4,6-三-O-叔丁氧基-2-脱氧葡萄糖基)丙基亚胺(22)作为手性模板,实现高选择性氰化,再经水解脱除辅助基团得(S)-23;随后与4-氯丁酰氯环化得(S)-24,再经95% H₂SO₄水解及酰胺化,获得目标产物1(92% yield)(Scheme 12)。但该路线因使用剧毒氰化物及强腐蚀性SnCl₄(用于亚胺活化),存在安全与设备兼容性风险,难以工业化放大。 Raju等改用Strecker反应:丙醛(25)与手性胺(S)-26缩合生成亚胺,CN⁻从位阻较小的re面进攻,立体选择性得到(1S, 2S)-27;水解得酸3,经SOCl₂/MeOH酯化、氨解得酰胺2,再与4-氯丁酰氯在碱性条件下环化得1(Scheme 13)。尽管步骤清晰,但因依赖NaCN且总收率仅42%,综合安全性与经济性,仍不适用于大规模生产。
2.2 不对称氢化
2-氧代丁酸(28)与2-吡咯烷酮(29)缩合得化合物30,再经酰氯化/胺化两步转化为脱氢左乙拉西坦(31)(Scheme 14)。化合物31通过不对称氢化制得目标物1:
— 方法A:以0.5 mol% (S,S)-Et-DuPhos/Rh(COD)₂OTf为催化剂,DCM为溶剂, 99%转化率,98% ee;
— 方法B:以0.08 mol% (R,R)-Ph-BPE/CoCl₂–Zn为催化体系,MeOH为溶剂,97%产率, 98.2% ee(Scheme 14)。
2.3 不对称氢化(以Nitrosobenzene作为原料)
Kotkar与Sudalai通过Nitrosobenzene的立体选择性插入、羟基化及Mitsunobu衍生化合成1即。丁醛(32)在L-脯氨酸催化下与Nitrosobenzene反应,还原得(R)-α-氨基氧醇(R)-33;再经Pd/C催化氢化(MeOH),得(R)-1,2-丁二醇(34)。(R)-34经Bu₂SnO/BrCH₂Ph选择性苄基化得(R)-35;其仲醇经MsCl甲磺酰化得(R)-36;后者与2-吡咯烷酮加热取代,高立体专一性地生成(S)-37。(R)-37脱苄基(H₂/Pd/C)后,经NaOCl–NaClO₂/TEMPO(cat.)氧化得酸(S)-9;再与氯甲酸乙酯和NH₃·H₂O缩合,以27%产率、>99.5% ee获得目标物1(Scheme 15)。
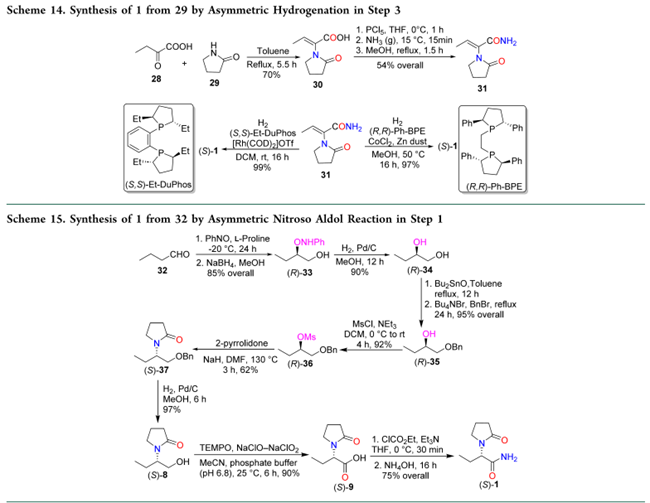
2.4 DYKAT策略
Imahori 等人利用动态动力学不对称转化(DYKAT)合成 1:环氧化物 38 与琥珀酰亚胺 39 在 π-烯丙基氯化钯二聚体/(R,R)-DACH(Trost 萘基配体)催化下反应,高选择性生成(S)-40;其羟基在 CF₃SO₃H 催化下与苯甲基三氯乙酰亚胺反应得(S)-41(NaH/BrCH₂Ph 法产率较低);(S)-41 经两步还原得酰胺(S)-42;后者在 H₂/Pd(OH)₂ 下氢化–氢解得醇(S)-8;再经 NaOCl–NaClO₂/TEMPO(cat.)氧化为酸(S)-9;最后与异丁基氯甲酸酯及 NH₃·H₂O 缩合,以 34% 总收率、>99% ee 获得 1(Scheme 16)。因需使用昂贵的(R,R)-DACH 配体,该路线不具工业可行性。
2.5 不对称乙基化
Chandra Babu 等人以 N-亚磺酰亚胺立体选择性乙基化合成 1:(R)-甘油醛缩酮((R)-46)与 (S)-叔丁基亚磺酰胺在 CuSO₄ 催化下缩合,构建手性模板 (S,E,S)-47;(R)-46 由 (R)-环氧氯丙烷经四步制得(31% 总收率)。EtMgBr 对 (S,E,S)-47 的 C=N 键进行椅式过渡态控制的 1,2-加成,高选择性生成 (S,S,S)-48;随后脱除 t-BuS(O)– 和缩醛保护基,得氨基二醇 (S,S)-49;NaIO₄ 氧化/NaBH₄ 还原得化合物 7;7 经 4-氯丁酰氯酰化、KMnO₄ 氧化得酸 (S)-9;(S)-9 氨解即得 1,但产率极低(<10%,Scheme 17)。
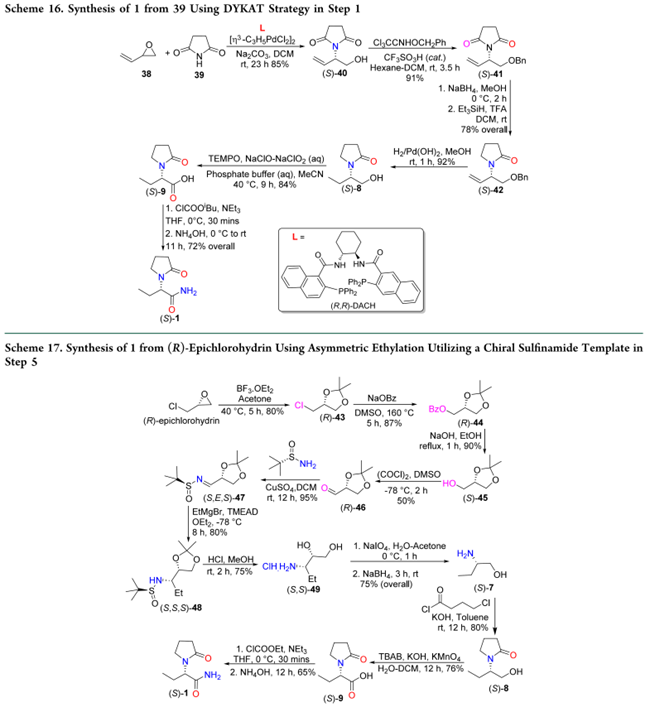
2.6 以钌催化剂(Grubbs催化剂)合成1
Liniger 等人通过烯烃化学选择性异构化–氧化串联策略合成 1:丙醛与 (S)-叔丁基亚磺酰胺缩合得手性亚磺酰亚胺 (S,E)-50;EtMgBr 误写,应为 AllylMgBr —— 已修正)经高度非对映选择性烯丙基化得 (S,E)-51;酸性脱除 t-BuS(O)– 辅助基得胺 (S)-52;再与 4-氯丁酰氯酰化得同烯丙基酰胺 (S)-53;随后在 Grubbs 催化剂作用下发生烯烃异构化,继而氧化生成酸 (S)-9;(S)-9 经氨解直接得到 1(67% 总收率,>98% ee,Scheme 18)。
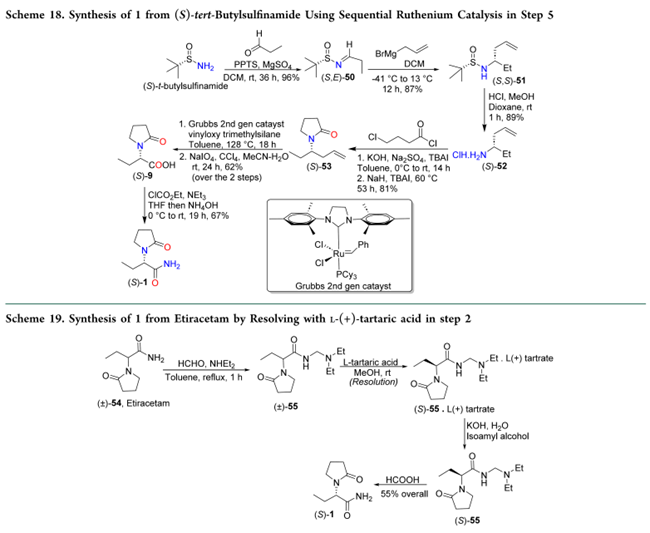
3、[拆分法]
3.1 手性拆分(±)Etiracetam
杨等29通过手性拆分(±)-乙酰拉西坦(54)制备(S)-左乙拉西坦(1):(±)-54与甲醛、二乙胺缩合得(±)-55;再以L-(+)-酒石酸选择性成盐,析出(S)-55的酒石酸盐,过滤、干燥后,用KOH中和得游离碱(S)-55;最后经甲酸处理,以55%总收率获得1(Scheme 19)。
3.2 手性拆分2-Aminobutyramide
利用 L-(+)-酒石酸拆分 2-氨基丁酰胺 ((±)-2) 得到 (S)-2-氨基丁酰胺 (2),其与 4-氯丁酰氯反应得到 1。从 2-溴丁酸 ((±)-56) 通过酰胺化反应可合成 (±)-2,将其与 L-(+)-酒石酸反应形成主要产物 S-(+)-2-氨基丁酰胺酒石酸盐,然后在碱存在下与 4-氯丁酰氯反应得到 1(Scheme 20)。
Boschi等人报道通过用(S)-N-苯基戊内酰胺作为手性辅助剂处理(±)-56 实现其消旋体拆分。其中(R,S)-57 是主要成分(67%,>98:2 dr),通过硅胶柱层析将其分离。使用 LiOH/H2O2 在 THF 中进行水解消除手性辅助剂,得到(R)-2-溴丁酸((R)-57)。在 THF 中以 NaH 为碱,用 2-吡咯烷酮对化合物 (R)-57 中的溴原子进行 SN2 取代,得到酸 (S)-9,其与氯甲酸乙酯和氨水反应,以 63% 的产率和>99% ee转化为 1。值得一提的是,用 N-苯基戊内酰胺拆分(±)-2-(2-氧代吡咯烷-丁酸和(±)-2-氯丁酸,在非对映异构体比方面结果不尽人意(Scheme 21)
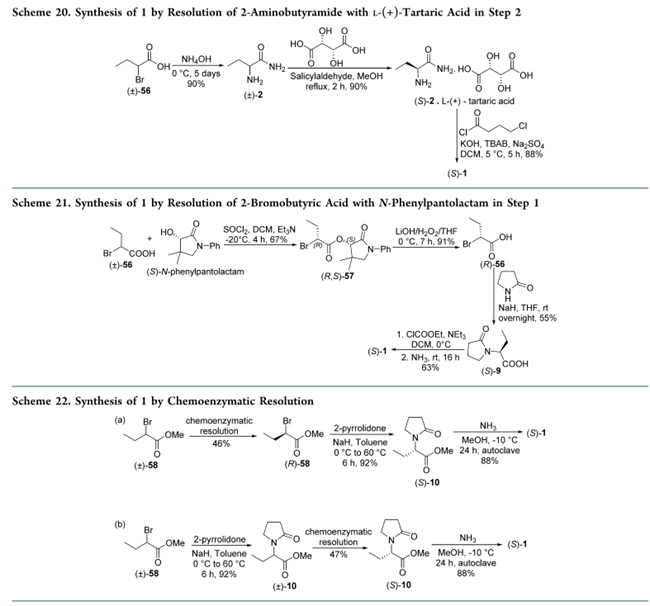
3.3 化学酶法拆分
Pan等利用固定化酶(Methylocystis spp,CCTCC M2016494)拆分消旋2-溴丁酸甲酯((±)-58),制备(S)-10,再经氨解得1。路线a(Scheme 22a):酶促拆分得(R)-58,随后在甲苯中以NaH为碱、2-吡咯烷酮进行SN2取代,高选择性生成(S)-10;路线b(Scheme 22b):先由(±)-58与2-吡咯烷酮发生SN2反应得(±)-10,再经拆分获得(S)-10。两条路线中,(S)-10均与氨反应定量转化为1。
3.4 (±)-2-(2-Oxopyrrolidin-1-yl)butyric Acid的拆分
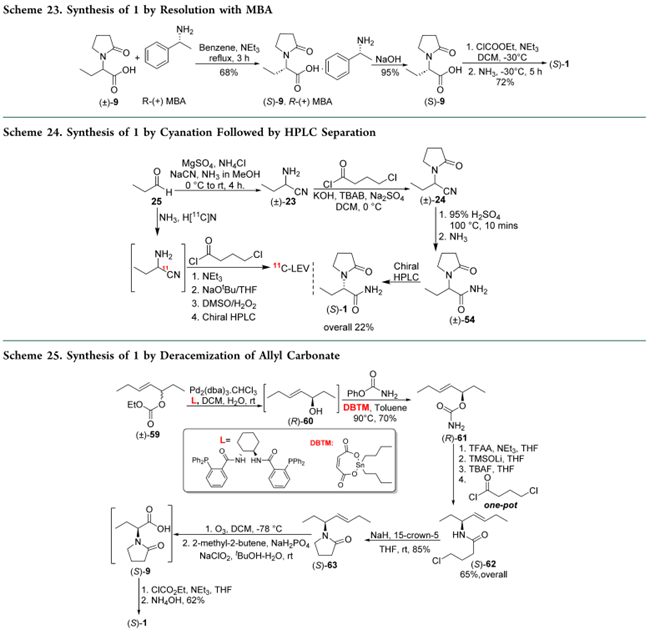
(±)-2-(2-Oxopyrrolidin-1-yl)butyric Acid((±)-9)在苯中、三乙胺存在下,与(R)-(+)-α-甲基苄胺(R-(+)-MBA)成盐拆分,析出(S)-9的R-MBA盐;碱处理后酸化得(S)-9;再经氯甲酸乙酯活化、氨解即得1(Scheme 23)。Neelakandan等改用甲苯/石油醚或THF替代苯,提升过程绿色性。
3.5 氰化和HPLC分离
Cai等以丙醛(25)为起始物:经氨缩合生成亚胺,再经Strecker氰化得(±)-23;与4-氯丁酰氯环合得(±)-24;95% H₂SO₄水解后酰胺化得(±)- etiracetam(54);手性HPLC分离后氨解,以22%总收率获得1(Scheme 24)。该路线亦用于一锅法放射性合成¹¹C-左乙拉西坦(¹¹C-LEV)。
3.6去外消旋化
Narczyk等采用Pd₂(dba)₃·CHCl₃/(R,R)-DACH-苯基配体的催化体系,对(±)-59进行动态动力学拆分,高选择性得(R)-烯丙醇60;未经纯化,直接锡催化转氨基甲酰化得(R)-61。后者经TFAA/Et₃N介导[3,3]-重排生成烯丙基异氰酸酯,再经TMSOLi/TBAF一锅处理得游离烯丙胺;随后与4-氯丁酰氯酰化得(S)-62,NaH促进环合生成(S)-63;臭氧化/Pinnick−Lindgren氧化得(S)-9;氯甲酸乙酯活化后一锅氨解,以24%总收率得1(Scheme 25)。
3.7水解动力学拆分
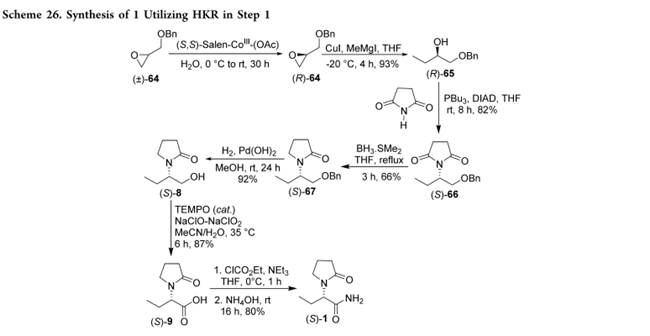
Muthukrishnan等利用HKR拆分外消旋苄基缩水甘油醚64,得对映体纯(R)-64;CuI/MgMeI区域选择性开环得(R)-65;Mitsunobu反应引入琥珀酰亚胺得(S)-66;部分还原构建2-吡咯烷酮得(S)-67;Pd(OH)₂氢解脱苄得(S)-8;TEMPO氧化得(S)-9;氯甲酸乙酯/氨解即得1(Scheme 26)。
总结:
本综述系统评述了(S)-左乙拉西坦(1)的各类合成路线及其优劣: 1)手性池法:以2-氨基丁酰胺、2-氨基丁酸或2-氨基丁醇为起始原料。优势是步骤少、路线短;缺点是存在部分消旋风险,并产生大量固体及酸性废液;2)天然氨基酸法:采用L-苏氨酸或L-甲硫氨酸。主要不足是步骤多、总收率低;3)不对称合成法:包括Trost DYKAT、不对称氰化、乙基化和氢化。受限于昂贵的手性配体、低温条件,以及氰化物、正丁基锂等高危试剂,难以工业化;4)拆分法:虽可行,但理论最大收率仅50%,造成显著物料损失。综合比较,以2-氨基丁酰胺为手性源的路线最具产业化潜力。
参考资料
A Short Review on the Synthetic Routes for the Antiepileptic Drug (S)‑Levetiracetam
Sayantan Paul and Asish K. Bhattacharya*
Org. Process Res. Dev. 2024, 28, 924−936. DOI: 10.1021/acs.oprd.4c00002


















No comments yet.