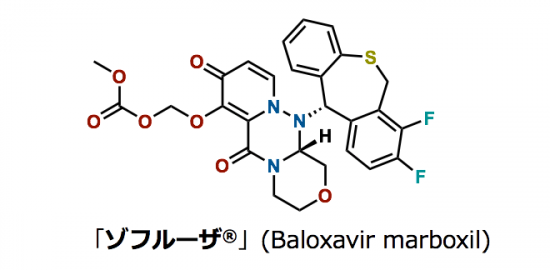
1小编正式进入实验室开始课题研究已经近三个月了,由于本科时进行过一些基础的科研训练,所以对于实验基本操作比较熟悉,然而实际进行实验的时候仍然有很多问题是在教科书中找不到答案的,需要自行摸索并积累经验,下面就将一些实用的小技巧分享给大家,欢迎大家进行尝试并总结一些更好的技巧方法。
一、称量的一些小技巧
1.反应往往需要的催化剂质量很小,有时不足1 mg,用天平称量不准(示数跳动)或者耗时过长,为了避免误差,可以将催化剂的量扩大倍数溶解于反应溶剂中,计算浓度后用注射器或者移液枪量取体积。
2.待称量的药品是较浓的油状液体,有时非常容易堵塞注射器针头,这时可以根据用量选择一个合适大小干净的离心管或者反应瓶,将其在天平上去皮置零,用滴管取出药品滴入离心管或反应瓶确定质量,当然取药品的滴管上肯定有残留的药品,如果药品非常昂贵不易得,我更推荐使用差量法称量:将药品瓶于天平上去皮置零,用滴管取药品,根据天平示数(这时会显示负数)得到自己需要的质量后将滴管一同放入干净的离心管或者反应瓶中,最后用溶剂涮洗几次以最大限度避免浪费。
3.有些药品室温下可能是液体,但是温度降低就会变成固体,有时这种药品称量前可以置于低温仪或-20℃冰箱冷冻一段时间,然后尽快的完成称量,否则就会在称量纸上变成液体。
二、特殊试剂的取用
此处所指的特殊试剂主要包括某些直接购买的干燥溶剂(底部含有分子筛)以及诸如格氏试剂、正丁基锂这样的活泼试剂。
1.对于第一种干燥溶剂,取用时没有特别要注意的。一般将试剂瓶先固定,在瓶口插上一个氮气球平衡内外压力(左图),先用注射器将瓶内气体吸出一部分,再用长针头注射器吸取即可。我有一个不错的习惯是新注射器在使用之前先用镊子把针头和注射器尖嘴拧一下,避免推动活塞时针头崩开发生危险。将针头从瓶口拔出时经常会出现液体滴落的情况,如果吸取液体之后将针头拔离液面稍稍吸进一部分氮气(右图),这样就可以解决此问题。还值得一提的是,有时试剂取用的人多,瓶盖会留下许多针孔,久而久之不利于试剂的干燥保存,因此取用完之后尽可能及时盖好外盖,最好能用封口膜密封一圈。

2.对于第二类试剂,由于试剂本身的特殊性,取用时应格外小心。大致操作和取用干燥溶剂类似,但是要格外注意取用完器具的淬灭,如果直接将注射器丢进垃圾桶,正丁基锂这种高度活泼的试剂很容易在空气中引起燃烧发生危险,通常是将取完的注射器直接丢入大量水中淬灭。还要注意的一点是这些试剂往往不是纯净的,都是溶解在某种惰性溶剂中,例如正丁基锂会溶解在正己烷中,浓度有1.6 mol/L和2.5 mol/L等,在计算体积时要格外注意。
三、NMR测试
核磁共振波谱是现代有机结构分析中最为重要的工具之一。当我们通过文献方法合成一个化合物时,需要将其投入下一步反应,在进行反应前最好通过氢谱确定结构是否正确,尤其是另一个反应物非常珍贵时;当我们自己在做未知反应的时候,从TLC上看到几个点,也得一个个分离后通过NMR确定结构,因此每一位有机合成工作者几乎每天都要和NMR打交道。
1.送样所需样品质量氢谱和碳谱差别很大,受限于13C在自然界的丰度以及灵敏度,碳谱用量往往多于氢谱,一般需要多于20 mg并合理选择扫描次数,较高浓度可缩短测试时间,基线也会平直。氢谱用量一般3-7 mg即可,浓度过大谱图的峰形以及裂分会不好,可能影响谱图解析和美观性;浓度过低则噪音较大、基线不平,往往还需要增加扫描次数。
2.NMR测试最理想的就是可以拿到“干净”的谱图。通常情况下我们拿到的谱图都会或多或少残留一些乙酸乙酯、二氯甲烷这样的非氘代试剂溶剂峰,在高场区往往还会出现硅脂(silicone grease)的峰,如果不作为文章发表这样的谱图尚可,一旦需要发表文章,“挑剔”的编辑就会对谱图质量有一定的要求。对于固体样品,往往比较容易拿到干净的谱图,固体样品通常不会像油状样品一样包裹溶剂分子,只要用油泵多抽一段时间,必要的时候可以用吹风机吹瓶子的底部(对热敏感的样品慎用),固体样品还可以考虑用小极性的石油醚重结晶的方法。油状液体较固体而言往往很容易包裹溶剂分子,有些化合物与乙酸乙酯结合非常紧密,即使在油泵下几个小时还会残留,而二氯甲烷则相对好一些,所以可以用二氯甲烷置换几次,甚至可以直接选择用氯仿进行置换。最后要提的是无论固体还是液体样品都会存在的硅脂峰,硅脂峰无论你如何小心都不能完全杜绝掉,因为实验用具的磨口、旋塞以及注射器的活塞等等在使用前就已经经过了特殊的处理,一般情况下只要信号不是特别强在处理谱图时是可以将其压在基线内的。如果信号强只能再进行纯化,比较简单的操作是用一根非常短的硅胶柱,上样后用石油醚或者正己烷淋洗50-100 mL,然后直接用乙酸乙酯将样品洗脱下来,这样大部分的硅脂都被小极性的溶剂冲掉了。
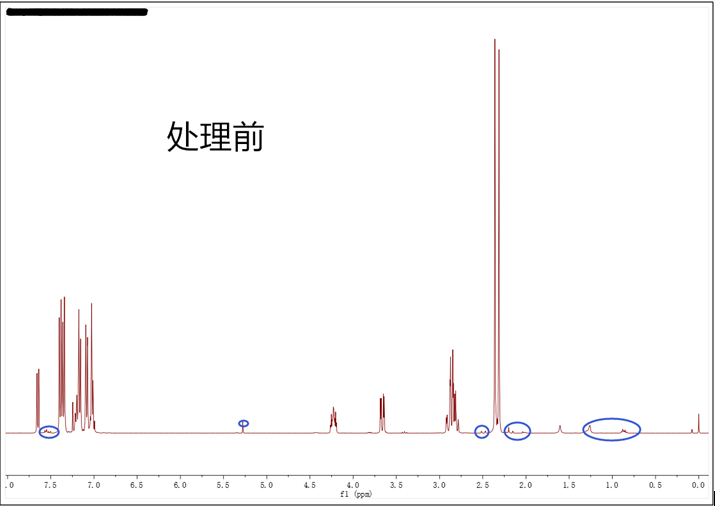
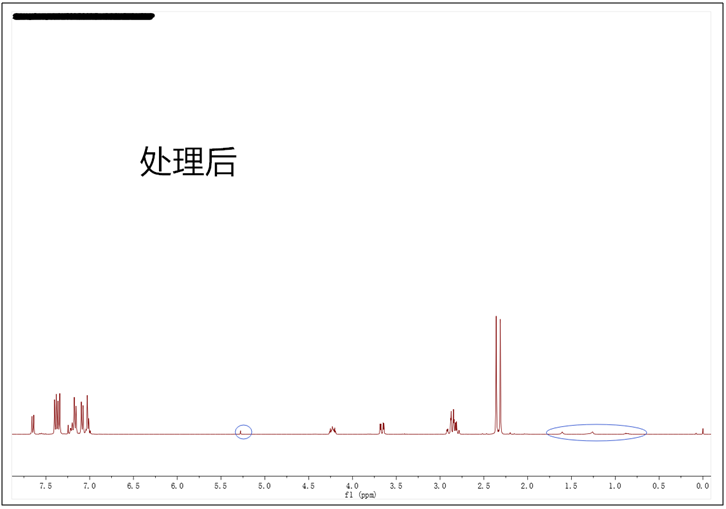
3.对于什么样的谱图是符合发表要求的,其实并无明确规定,各个期刊以及不同的编辑和审稿人的标准也不一样。在处理谱图时也有一些技巧,以常用的MestReNova软件为例,如果化合物的浓度比较大,峰也会比较高,杂质峰就会很矮,我们通过鼠标滚轮将峰压低,这时一些小的杂质就会被“藏”进基线内。例如下图小编合成的一个化合物,打开软件发现谱图比较干净,但还是存在一些小的杂质和5.30 ppm处的DCM残留溶剂峰(已用蓝色圈出),事实上2.4 ppm处的两个甲基峰非常高,处理谱图时适当压低就会得到最理想的状态。另外处理谱图时,我们也要选择合适的字体、大小以及颜色,确保积分值、标峰能够清楚明了,还要将结构式画在谱图空白的地方,必要的时候最好放上局部放大图突显严谨性。
纸上得来终觉浅,以上仅仅是小编实验过程中总结的一些小tips,不一定适合每个人也不一定完全规范和奏效,只有在实验室多观察多尝试多总结才会有意外收获,希望各位化学人儿都能实验顺利!
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!