
本文作者:杉杉
导读
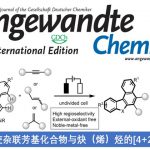
近日,上海有机所游书力课题组在Angew. Chem. Int. Ed.中发表论文,报道了钯催化的3-硝基吲哚底物与烯丙基碳酸酯 (allyl carbonates)之间的去芳构化甲氧烯丙基化 (dearomative methoxyallylation)反应方法学。该方法学具有底物应用范围广泛、较高的反应收率 (高达86%)与良好的非对映选择性 (>20:1 dr)等优势。同时,在克级规模实验的研究中,作者发现,仅需要采用较低的催化剂负载量 (1 mol%),便能够有效地完成相应的去芳构化甲氧烯丙基化过程,进而表明这一方法学的具有较好的合成实用性。此外,动力学实验研究表明,烷氧负离子的亲核进攻为反应过程中的决速步骤。
Palladium-Catalyzed Dearomative Methoxyallylation of 3-Nitroindoles with Allyl Carbonates
J. Xie, C. Zheng, S. You, Angew. Chem. Int. Ed.ASAP. doi: 10.1002/anie.202107139.
正文
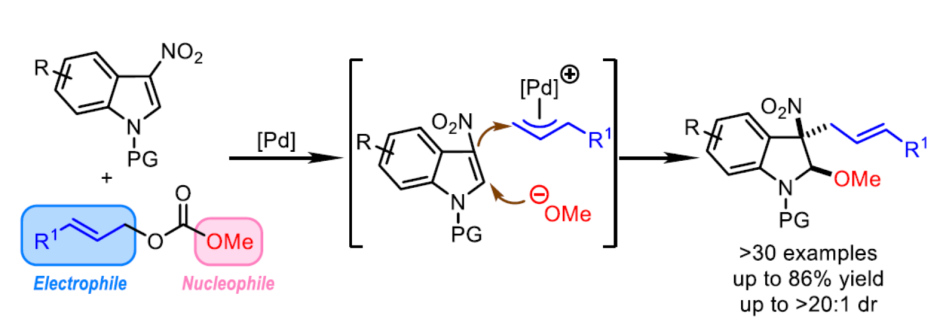
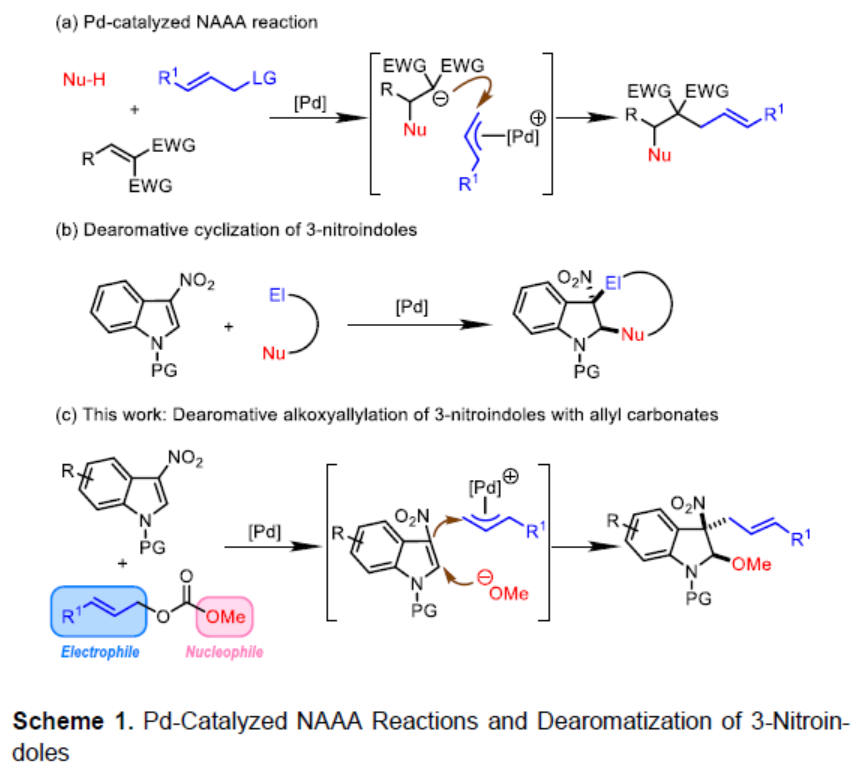
钯催化的烯丙基取代反应方法学已经成为当代有机合成化学中构建C-C或C-X 键(X=O、N、S等)最为有效的方法之一。然而,在上述的合成转化方法学中,多数反应过程,仅能手性一种类型的共价键的构建。其中,亲核加成诱导的烯丙基烷基化 (nucleophilic-addition-induced allylic alkylation, NAAA)方法学[1]的研究则更加具有吸引力,其反应过程涉及如下步骤:首先,通过外加亲核试剂对Michael受体的进攻进攻,产生新的亲核试剂,接下来,新形成的亲核试剂再与亲电性的π-烯丙基-Pd配合物进行后续的反应步骤 (Scheme 1a)。通过NAAA方法学,能够连续地形成两种不同类型的化学键,并具有优良的可控性,同时,能够实现可极化型C=C双键的双重官能团化 [2]。然而,上述NAAA反应方法学中,Michael受体的应用范围在较大程度上局限于亚苄基丙二腈及其类似物,进而使NAAA方法学策略的进一步应用受到较大限制。近年来,催化去芳构化反应方法学备受关注,该方法学能够将平面结构的芳香底物直接转化为一系列相应的环状化合物[3]-[9]。目前,通过分子内或分子间亲核加成引发的吲哚去芳构化反应方法学已有大量文献报道[10]-[15]。同时,缺电子吲哚的去芳构化策略作为吲哚去芳构化方法学的一种互补策略,同样能够应用于一系列多重官能团化的二氢吲哚分子的构建[16]。尤其是3-硝基吲哚底物与一系列两亲性试剂 (amphiphilic reagents)之间的去芳构化成环反应方法学的发展,最终,能够将亲核试剂与亲电试剂分别引入吲哚环的C2-与C3-位置 (Scheme 1b) [16]。
受上述研究的启发,作者设想,能否将NAAA策略应用于钯催化的3-硝基吲哚底物与烯丙基碳酸酯之间的去芳构化反应过程 (Scheme 1c)。其中,通过烯丙基碳酸酯与钯催化剂作用,同时产生具有亲核性的甲氧基负离子与具有亲电性的π-烯丙基-钯配合物,并分别在3-硝基吲哚的C2-与C3-位置进行亲核与亲电进攻,从而获得预期的去芳构化产物。并且,与已报道的3-硝基吲哚底物去芳构化成环反应相比,设计这一策略的主要挑战在于在预期的去芳构化反应过程中,如何调节三种反应参与物之间的反应活性差异。这里,作者进一步设想,在预期的去芳构化反应过程中,甲氧基负离子 (硬亲核试剂)优先进攻3-硝基吲哚 (硬Michael受体)。相反,原位形成的碳负离子 (软亲核试剂)更容易优先进攻π-烯丙基-钯配合物 (软亲电试剂)。
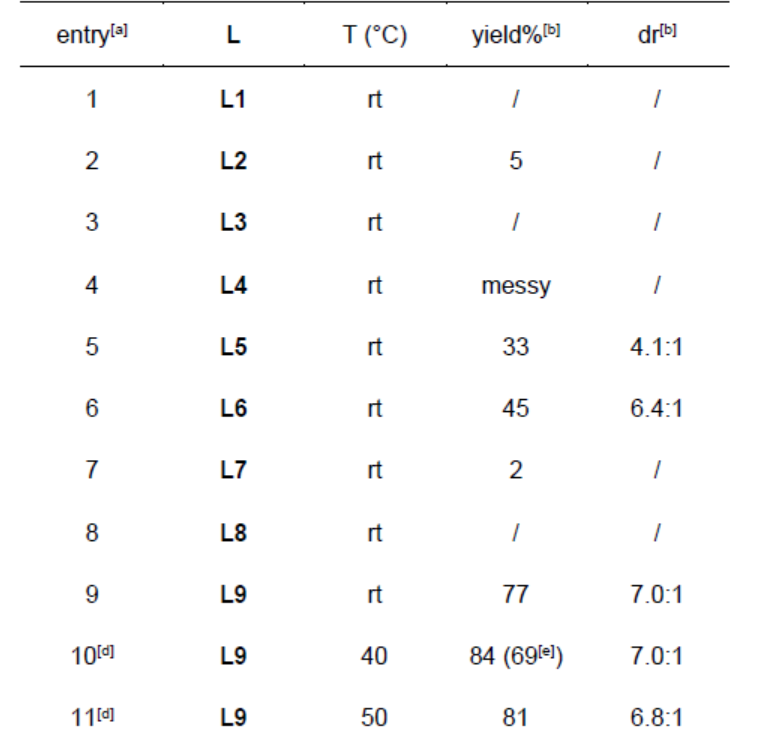
首先,作者采用N-叔丁氧羰基-3-硝基吲哚1a与烯丙基甲基碳酸酯2a作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用5 mol% Pd2dba3作为催化剂,11 mol% L9作为配体,乙腈作为反应溶剂,反应温度为40 ℃,最终,能够获得84%收率的3aa,d.r.为7:1 (entry 10)。


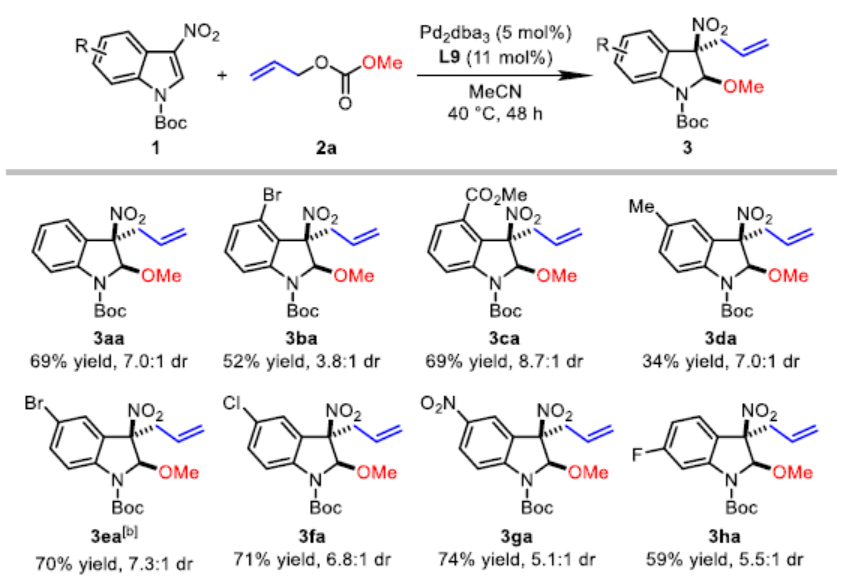
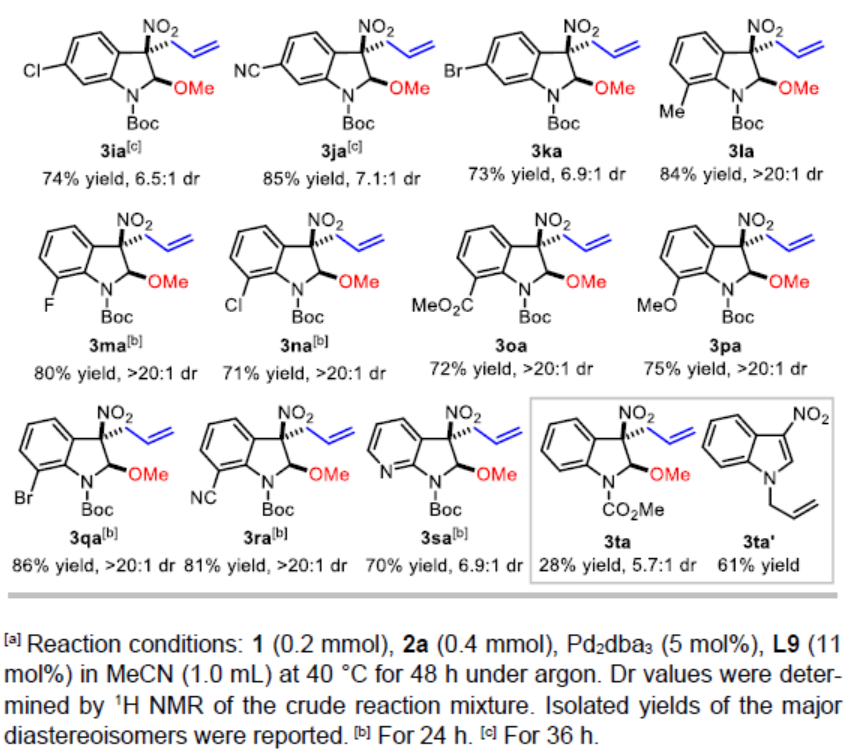
在上述的最佳反应条件下,作者对3-硝基吲哚的底物应用范围进行考察(Table 2)。研究发现,在吲哚环的C4-、C5-或C6-位带有吸电子基团 (CO2Me、F、Cl、Br、NO2、CN)取代的3-硝基吲哚底物,均能够获得相应的产物3ba、3ca以及3ea–3ka,收率为52-85%,dr为3.8:1-8.7:1。同时,作者发现,在吲哚环的C5-位中带有供电子的甲基基团取代时,可能由于底物具有较弱的亲电性,因此,使产物3da的收率显著降低 (34%)。值得注意的是,上述的反应条件对于7-取代的3-硝基吲哚底物,同样能够获得具有优良非对映选择性 (dr>20:1)的产物3la–3ra,反应收率为71-86%。而且,带有溴取代基的吲哚底物,同样能够与上述的Pd催化条件良好地兼容 (3ba、3ea、3ka以及3qa)。此外,7-氮杂吲哚底物同样能够有效地参与上述的去芳构化甲氧烯丙基化反应过程,并以70%的收率获得相应产物3sa,dr为6.9:1。最后,作者观察到,N-Boc保护基对于上述反应过程的顺利进行尤为重要。采用N-CO2Me保护的底物,在上述标准条件下进行相应的去芳构化反应过程,则仅能够获得28%收率的产物3ta,即最终的反应收率出现显著降低。并同时获得副产物1-烯丙基-3-硝基-1H-吲哚 (3ta’),收率为61%。此外,实验发现,对于其它N-保护基团 (例如Ts与Bn)取代的吲哚底物,均无法有效地参与上述的去芳构化甲氧烯丙基化过程。
![]()
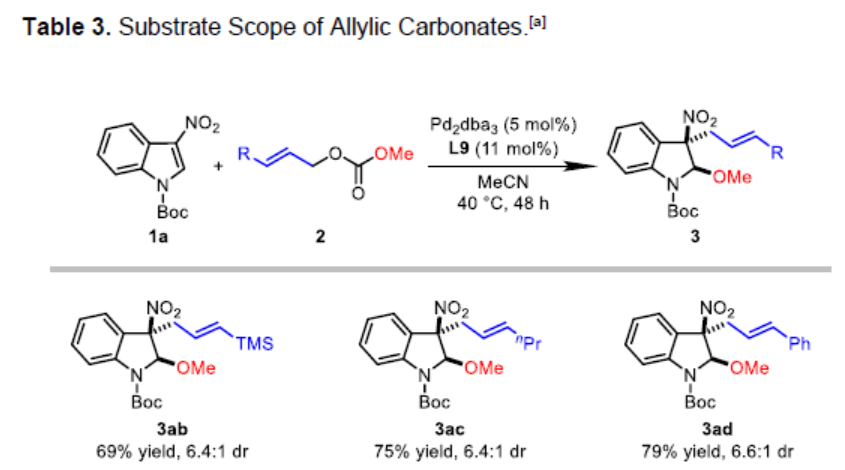
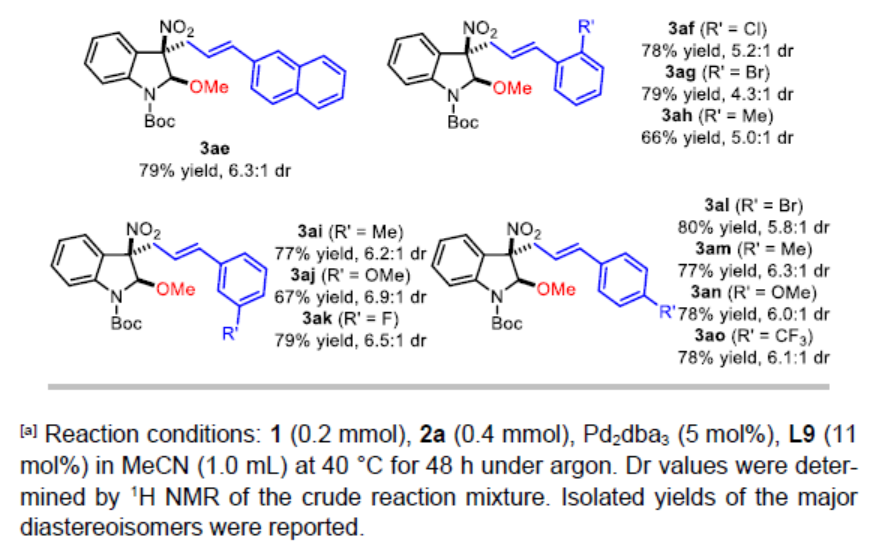
接下来,作者对烯丙基碳酸酯的底物范围进行进一步考察 (Table 3)。研究表明,具有三甲基甲硅基或丙烯基取代的烯丙基碳酸酯底物,能够以中等程度的反应收率 (69%与75%)与良好的非对映选择性 (dr为6.4:1)获得相应产物3ab与3ac。同时,作者发现,一系列在芳基不同位置带有供电子或吸电子基团取代的芳基烯丙基碳酸酯底物,均能够与上述的标准反应条件良好的兼容,并获得预期产物3ad–3ao,收率为66-79%,dr为4.3:1-6.3:1。值得注意的是,邻位取代的芳基烯丙基碳酸酯底物在参与上述的去芳构化反应过程时,非对映选择性略有降低 (3ag、3ah与3ai、3al以及3am),这可能源自于π-烯丙基-钯配合物参与亲核进攻时,过渡态中存在显著的立体阻碍。
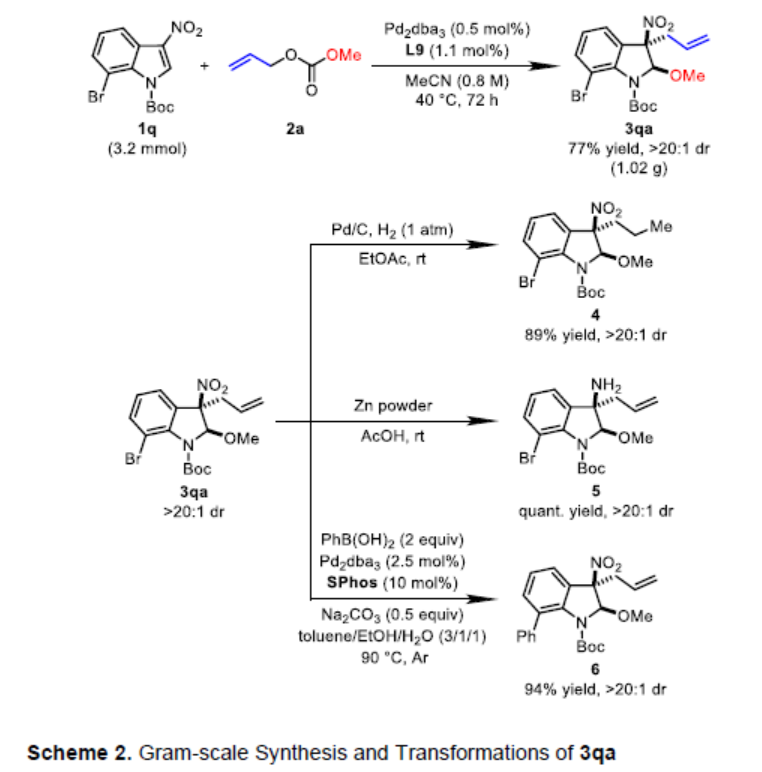
之后,作者对上述去芳构化甲氧烯丙基化方法学的合成实用性进行相关研究 (Scheme 2)。实验结果表明,将底物1q的用量扩大至3.2mmol时,选择更长的反应时间 (72 h)与较低的催化剂负载量 (1 mol%),同样能够获得77%收率产物3qa,dr>20:1。同时,作者对相应的去芳构化甲氧烯丙基化产物3qa进行一系列不同类型的合成转化研究。首先,将3qa采用Pd/C进行末端C=C键的氢化反应,能够获得89%收率的产物4。其次,3qa中的硝基官能团能够通过Zn粉/HOAc体系,进一步还原为胺 (5)。此外,3qa中的溴取代基能够有效地参与Suzuki-Miyaura交叉偶联过程,并获得94%的收率偶联产物 6。
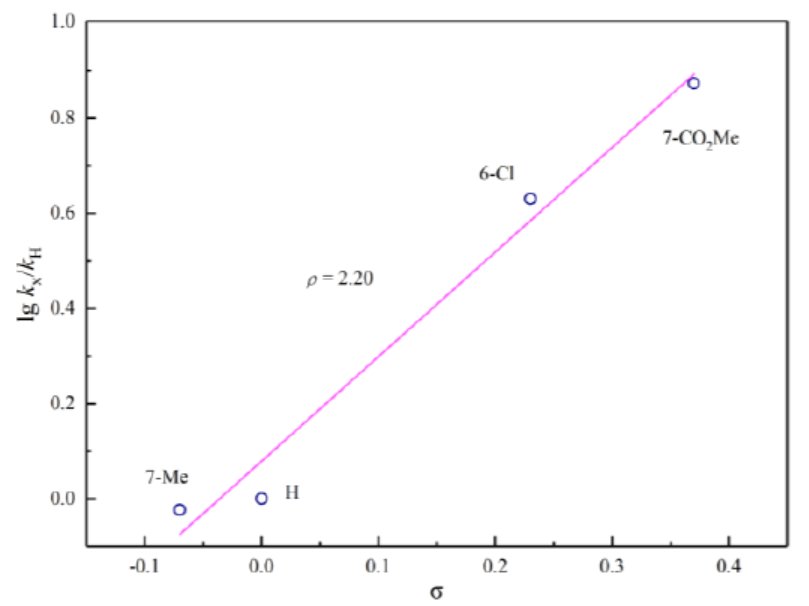
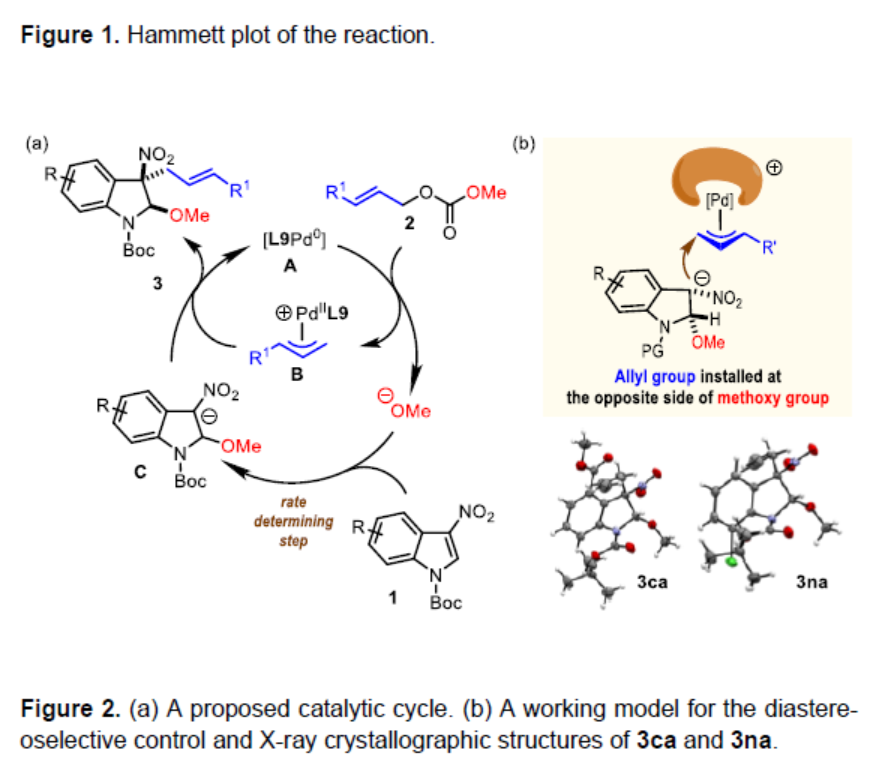
为阐明合理的反应机理,作者通过1H-NMR对一系列3-硝基吲哚底物进行相关的Hammett分析 (Figure1)。实验过程中,观察到正ρ值 (2.20),表明在决速步骤中存在负电荷的积累。基于这一实验观察,作者提出一种合理的反应机理 (Figure 2a)。首先, Pd(0)催化剂A与烯丙基碳酸酯2进行配位。接下来,Pd(0)经历氧化加成步骤,形成π-烯丙基-Pd(II)中间体B,并释放出甲氧基负离子。通过甲氧基负离子对3-硝基吲哚的C2-位的亲核进攻,形成关键的中间体C,中间体C中,吲哚环的C3-位出现形式负电荷。根据Hammett plot的分析,表明C2-位的亲核进攻可能为反应过程中的决速步骤。最终,通过中间体C对中间体B的亲核进攻,获得预期的去芳构化产物,并再生Pd(0)催化剂。此外,根据3ca与3na相对构型的研究,作者设想,烯丙基优先进攻二氢吲哚环中与甲氧基官能团位于相反一侧的反应位点,这源自于π-烯丙基-钯配合物进行亲核进攻的过渡态中,立体位阻的最小化 (Figure 2b)。
总结:上海有机所游书力课题组报道了钯催化的3-硝基吲哚底物与烯丙基碳酸酯之间的去芳化甲氧烯丙基化反应,从而以较高的反应收率以及高度的非对映选择性,获得一系列双重官能团化的二氢吲哚衍生物。同时,通过克级规模合成实验以及产物后期的衍生化过程,进一步阐明上述去芳构化反应方法学的合成应用价值。接下来,作者通过Hammett分析表明,甲氧基负离子对3-硝基吲哚的亲核进攻为反应过程中的决速步骤。
参考文献:
[1] H. Nakamura, M. Sekido, M. Ito, Y. Yamamoto, J. Am. Chem. Soc. 1998, 120, 6838. doi: 10.1021/ja980959a. [2] a) H. Nakamura, J.-G. Shim, Y. Yamamoto, J. Am. Chem. Soc. 1997, 119, 8113.doi: 10.1021/ja971599e. b) H. Nakamura, K. Aoyagi, J.-G.Shim, Y. Yamamoto, Am. Chem. Soc. 2001, 123, 372. doi: 10.1021/ja002931g. [3] C. Zhuo, W. Zhang, S. You, Angew. Chem. Int. Ed. 2012, 51, 12662. doi: 10.1002/anie.201204822. [4] Zhuo, C. Zheng, S. You, Acc. Chem. Res. 2014, 47, 2558. doi: 10.1021/ar500167f. [5] C. Zheng, S. You, Chem. 2016, 1, 830. doi: 10.1016/j.chempr.2016.11.005. [6] J. An, M. Bandini, Chimia 2018, 72, 610. doi: 10.2533/chimia.2018.610. [7] J. Bariwal, L. G. Voskressensky, E. V. Van der Eycken, Chem. Soc. Rev. 2018, 47, 3831. doi: 10.1039/C7CS00508C. [8] F. Sheng, J. Wang, W. Tan, Y. Zhang, F. Shi, Org. Chem. Front. 2020, 7, 3967. doi: 10.1039/D0QO01124J. [9] C. Zheng, S. You, ACS Cent. Sci. 2021, 7, 432. doi: 10.1021/acscentsci.0c01651. [10] Roche, J. Y. Tendoung, B. Tréguier, Tetrahedron 2015, 71, 3549. doi: 10.1016/j.tet.2014.06.054. [11] G. Mei, X. Tang, Y. Tasdan, Y. Lu, Angew. Chem. Int. Ed. 2020, 59, 648. doi: 10.1002/anie.201911686. [12] H. Chu, J. Cheng, J. Yang, Y. Guo, J. Zhang, Angew. Chem. Int. Ed. 2020, 59, 21991. doi: 10.1002/anie.202010164. [13] L. Lombardi, D. Bellini, A. Bottoni, M. Calvaresi, M. Monari, A. Kovtun, V.Palermo, M. Melucci, M. Bandini, Chem.–Eur. J. 2020, 26, 10427. doi: 10.1002/chem.202001373.
[14] W. Wu, L. Ding, L. Zhang, S. You, Org. Lett. 2020, 22, 1233. doi: 10.1021/acs.orglett.9b03988. [15] Z. Fu, J. Zhu, S. Guo, A. Lin, Chem. Commun. 2021, 57, 1262. doi: 10.1039/D0CC07529A. [16] a) A. Cerveri, M. Bandini, Chin. J. Chem. 2020, 38, 287. doi: 10.1002/cjoc.201900446.b) B. Rkein, A. Bigot, L. Birbaum, M. Manneveau, M. De Paolis, J.Legros, I. Chataigner, Commun. 2021, 57, 27.doi: 10.1039/D0CC06658C. [17] a) B. M. Trost,V. Ehmke, B. M. O’Keefe, D. A. Bringley, J. Am. Chem. Soc. 2014, 136, 8213. doi: 10.1021/ja5044825.b) J. Zhao, Z. Wu, M.Zhou, X. Xu, X.. Zhang, W. Yuan, Lett. 2015, 17, 5020. doi: 10.1021/acs.orglett.5b02489.













No comments yet.