
本文投稿作者 大白菜
(封面图片及文中图片均来自论文)
摘要
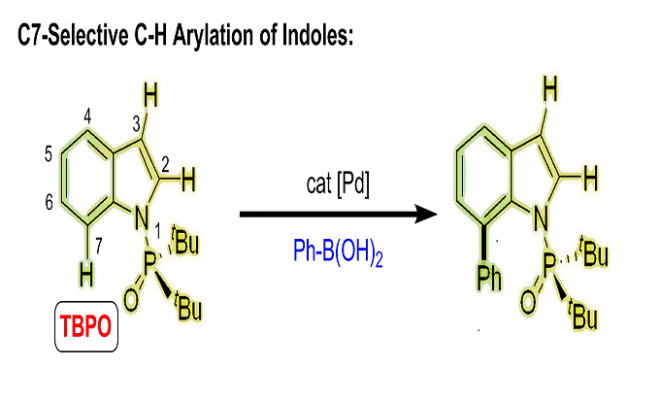
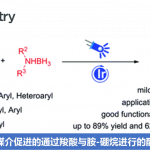
南京大学史壮志课题组(主页)报道了Pd催化吲哚与芳基硼酸的偶联反应可以高选择性引导至C7位的C-H活化过程,这种高度区域选择性的关键是在Pd(OAc)2催化下选择适当的膦酰基导向基团和吡啶类配体,尽管文章发表了一段时间,我们拿出来给大家介绍一下,。
Palladium-Catalyzed C−H Arylation of Indoles at the C7 Position
Youqing Yang, Xiaodong Qiu, Yue Zhao, Yucheng Mu, and Zhuangzhi Shi
J. Am. Chem. Soc. 2016, 138, 495−498, DOI: 10.1021/jacs.5b11569
研究背景
吲哚基团是具有生物活性天然产物的普遍特征,也是药物应用的重要结构。因此,开发有效的吲哚类区域选择性功能化方法受到了高度关注。在吲哚核心的N原子中心周围是一个邻位(C2)和两个可以官能化的间位(C3和C7)。通常情况下,吲哚的金属化和C-H活化发生在C3位置。为了克服固有的选择性,在N上引入像乙酰基,新戊酰基,N,N-二甲基氨基甲酰基和嘧啶基的导向基(DG)已经成为保证C2位选择性的有效策略(Scheme 1a)。形成鲜明对比的是,直接选择在C7位活化的方法仍然很少。通常,吲哚的C7位的选择性官能化需要取代C2位以阻断该位点处的反应性,或者,另外的有效途径包括将吲哚还原成相应的二氢吲哚衍生物,然后是区域选择性C-H官能化的二氢吲哚衍生物,随后氧化,可提供各种C7选择产物。因此需要一种能够在C7位置直接进行吲哚的C-H芳基化的新方法。
吲哚C7位选择性的挑战来自于在C2位置通过C-H键断裂形成五元素金属环优于在C7位置形成相应的六元金属环。作者推测,通过在N原子上使用庞大和更吸电子的DG,可以提升吲哚的C7位选择性,因为C7位置比C2位置空间位阻更大,电子差更小。接着作者考察了一系列具有不同空间体积的膦酰基DGs(5-8a)(如图scheme 1b)。
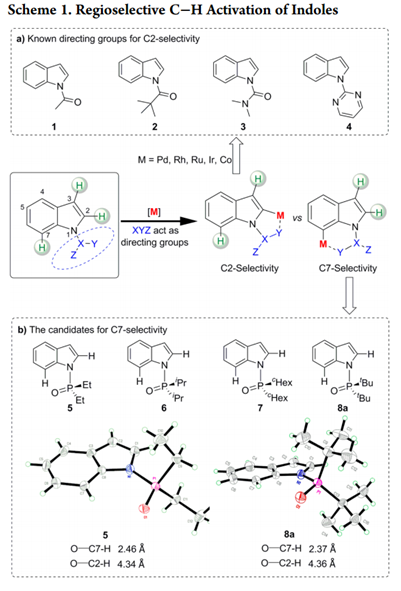
X射线晶体结构显示,在固态下,5(R = Et)和8a(R = tBu)中的O原子完美地面向C7位C−H活化的位置。我们推测酰胺N-P键可以在高温溶剂中自由旋转,导致C2和C7选择性差。来自8a中二叔丁基取代基的空间位阻可能会增加酰胺N-P键旋转的活化能,导致高度限制O−C7−H和O−C2−H构象之间相互转换。
最优反应条件优化
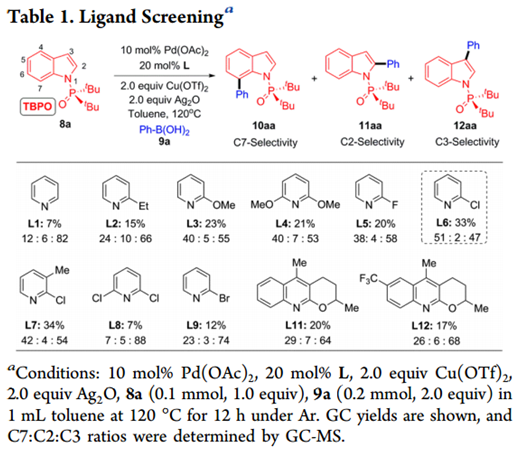
作者首先以二叔丁基-(1H-吲哚-1-基)氧化膦(8a)和苯硼酸(9a)作为模板底物进行尝试(如Table 1所示)。以10mol%Pd(OAc)2为催化剂,以20mol%吡啶为配体,在120℃下,2.0当量Cu(OTf)2和2.0当量Ag2O作为甲苯中的氧化剂,我们确实观察到中等产率的C7-,C2-和C3-芳基化产物10aa-12aa的混合物,尽管C7选择性比较低 (C7:C2:C3 = 12:6:82)。C7比C2更高的选择性表明该DG可以忽略C2位置的导向效应。作者接下来研究了各种取代的含N杂环(L2-L12)作为可能潜在地增加C7选择性的配体。其中,2-氯吡啶(L6)似乎具有最佳的空间平衡和电子性质,并以极好的C7(C7:C2 = 51:2)位选择性和33%收率得到10aa。
使用最佳配体L6,我们筛选出其他因素来抑制C3选择性(如图Table 2所示)。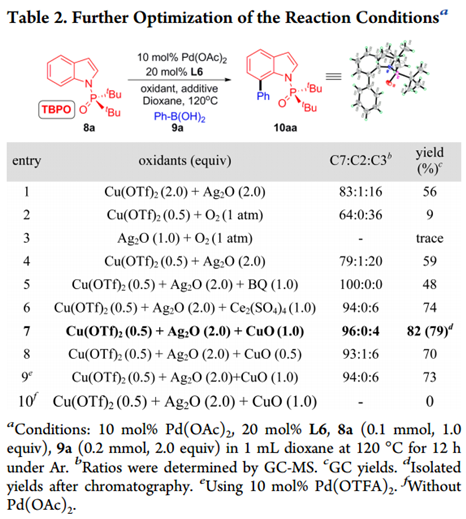
将溶剂转化为二氧六环使产率和C7选择性显着提高(Entry 1)。Cu和Ag盐联用是必需的(Entry 2,3),0.5等量Cu(OTf)2足以产生类似的结果(Entry 4)。经过广泛的筛选,作者发现添加第三种辅助氧化剂可以改善结果(Entry 5-7)。加入CuO有最明显的效果,10aa的产率显着提高到82%,具有优异的C7选择性(C7:C2:C3 = 96:0:4)。在这些条件下,减少CuO量或改变Pd源都会导致产率降低(Entry 8,9)。对照反应证实在不存在Pd(OAc)2的情况下反应无法进行(Entry 10)。
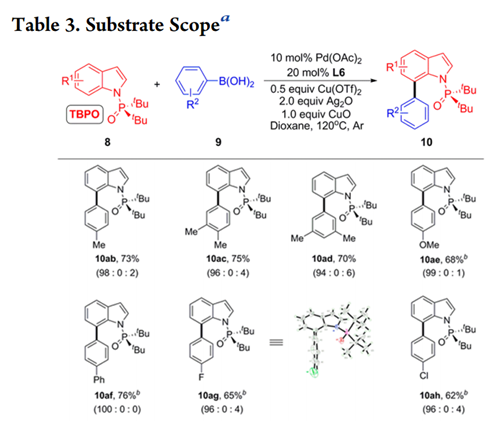
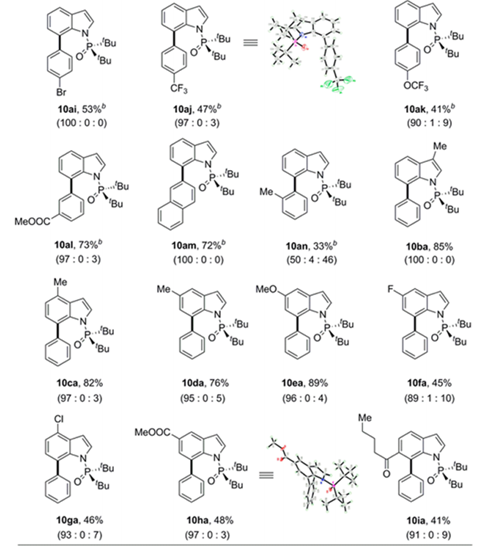
底物拓展
得到最优条件后,作者考查了C-H芳基化反应的适用范围(Table 3)。首先研究了8a与广泛的富电子和缺电子的芳基硼酸的交叉偶联反应,在芳环间位和对位带有甲基的苯硼酸以70-75%的产率提供所需产物,具有优异的C7选择性(10ab-10ad)。作者还将底物拓展到其他基团取代的吲哚,都能以41%—82%的产率得到产物,并具有一致的高区域选择性。
为了确定空间位阻P(O)tBu2部分对于C 7高选择性的重要性,作者在优化的反应条件下进行了对照实验(Table 4)。

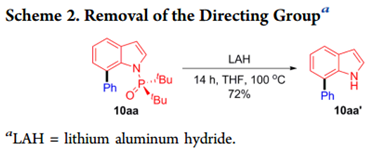
含有N-引导基团(如1-4)的吲哚提供了预期的C2和C3芳基化产物的混合物。接下来作者评估了取代基的位阻对膦酰基的影响,观察到底物带有P(O)Et2(5)和P(O)iPr2 (6)基团的吲哚不能形成任何C7位选择性产品,但包含P(O)cHex 2部分的底物(7)的C-H芳基化提供了34%收率的C7位选择性的所需产物。这些结果表明,尽管配体L6可以促进C7选择性,空间位阻的P(O)t Bu 2部分是实现高水平区域选择性的先决条件。通过用氢化铝锂处理10aa可以很容易地除去N-P(O)tBu 2 DG,得到72%产率的未保护的吲哚10aa’(Scheme 2)。
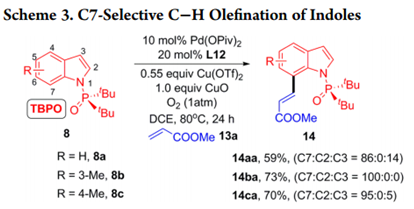
这个发现也能使用P(O)tBu2部分用于其他类型的交叉偶联,吲哚底物8a进行氧化Heck反应。作者发现使用丙烯酸甲酯(13a)作为
偶联剂与吲哚8a-8c在 P(O)Piv2作为催化剂, L12作为配体的存在下以59-73%的产率得到具有优异的C7区域选择性的C-H烯化产物14aa-14ca(Scheme 3)。
反应机理
为了深入了解该反应的机制和区域选择性,作者进行了氘化实验。仅在不存在9a时用D2O进行反应才在C3位置发生显着的氘化(Scheme 9a),在2.0当量9a的存在下也进行相同的实验,并在12小时后停止, 分离出21% D3-10aa和72% D3 / D7-8a (Scheme 9b)。
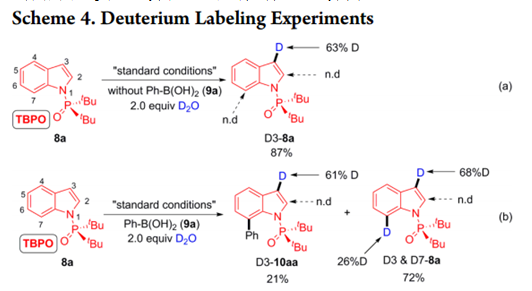
这些结果表明,最初的二价钯可以首先与苯硼酸转金属化[14],然后在P(O)tBu2辅助下发生8a C7位置的C-H活化。
总结
总之,史壮志课题组首次报道了在空间位阻和可移除的N- P(O)tBu2基导向下,Pd(II)催化吲哚的C7选择性C-H芳基化。这种新型的催化体系可以消除邻位效应以及在吲哚C2和C3位置产生电子偏差。这些现有结果代表了一个重要的发现,预计能够在吲哚的高度C7选择性C-H官能化中可以扩展到其他体系。发展新的交叉偶联反应和机理研究是史壮志课题组正在进行的研究课题。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!

















No comments yet.