译自Chem-Station网站日本版 原文链接:Gaussian Input File データベース
翻译:炸鸡
你喜欢计算化学?喜欢用Gaussian软件吗?笔者我就是计算化学爱好者!量子化学计算把看不见的电子微观世界活灵活现地展现在我眼前,冲它这一点我就果断爱上了它~。除了荧光分子的相关研究外,背靠量子化学计算的轨道计算就算是化学研究必不可少的工具了。
爱归爱,可有时它却让我很难弄。我所在的实验室虽然计算设备很完备,但很少有同僚们自己鼓捣计算。即使是做最熟悉的泛函数和基函数计算,短短几分钟里写个input文件然后投入Gaussian里指令计算也充满了波折与变数。
化学空间曾刊登过《从最简单的反应开始~量子化学计算求解中间体能量值~》的一篇简单的计算化学入门迷你教程,为了更好地帮助想学习计算化学的同学们,这里我将提供一些常用的Gaussian input file的模板。
使用Gaussian软件的量子化学计算流程
首先先简略看一遍用Gaussian做计算的流程。本文内容称不上是业内宝典名师权威,但对于那些没有充分了解量子化学但还想做量子化学计算的同学们来说是个很好的指导教程!(当然,同时量子化学的学习请不要松懈)
我观察了我周围的情况后发现,很多委托别人来做Gaussian计算的人都是出于担心使用软件需要数学知识和计算能力,自己不能胜任的心理所以选择交给其他人来做。
当然理解计算和数学原理是最好的,但不妨先姑且学学怎么用这个软件。
Gaussian的计算流程十分简单:
- 首先创建分子的结构信息(.xyz﹑笛卡尔坐标或z-matrix),可以在Gauss View, Mercury, Avogadoro, Chem3D等软件上画出分子的结构。
- 创建Gaussian Input file (.gjf)(在text edit或Gauss View上操作)
- 向Gaussian提交Gaussian Input file(在Gaussian上操作)
- 验收・分析Gaussian Output file (文件后缀名为.chk/.fchk/.log),可以在text edit, Gauss View, iqmol上操作
本文的全部内容都是关于整套流程中最重要的第二步——创建Gaussian Input file (.gjf)。Gaussian Input file 实质上是一种文本文档,它汇总了我们想让Gaussian做什么样的计算的指令信息,这些指令信息在Input file上为软件可以读取的形式。我曾幻想过有一天某位神人能打造出一个完美的Input file,完美到让Gaussian直接解出薛定谔方程,那量子化学界也就迎来了它生命的终点,当然这是不可能的。
那么,Input file该怎么写呢?通过在 Gauss View的操作,Input file可以自动生成,想要完美的Input file可以打开Input file的Text形式,在Text上修改。虽说规则都在HPC手册和Gaussian官方网站。但没有示例很难自己写可怎么办呢?不如到这篇文章里找找模板吧。只需将本文里的模板直接复制粘贴到text file里面就好(因为格式差异,可能有些空行的长短有错误,还请读者见谅)。修改分子的XYZ轴的信息和最开始的两行,最后加上两行的空行差不多就可以投入到Gaussian里进行计算了。(如有错误,欢迎读者指正)
例1) 以b3lyp/6-31g(d,p) 为泛函/基组的基态计算(S0构造最优化,振动数计算)
%nprocshared=28
%mem=100GB
%chk=TNT.chk
# opt freq b3lyp/6-31g(d,p)
****
0 1
C -4.59255 0.08304 -0.05852
C -3.07318 0.17974 -0.06585
C -2.35822 1.43873 -0.06606
N -2.97016 2.74777 -0.05954
O -2.21637 3.82816 -0.06039
O -4.14959 2.90090 -0.05332
C -0.94837 1.40993 -0.07316
C -0.22055 0.22236 -0.07994
N 1.20996 0.24774 -0.08700
O 1.85923 1.39310 -0.08718
O 1.83665 -0.76914 -0.09283
C -0.91178 -0.98707 -0.07972
C -2.31693 -1.04992 -0.07289
N -2.91158 -2.36710 -0.07344
O -2.14448 -3.43785 -0.08011
O -4.09243 -2.53018 -0.06800
H -5.18863 0.99767 -0.05309
H -4.91946 -0.45909 0.85345
H -4.92857 -0.45422 -0.97006
H -0.39416 2.33820 -0.07345
H -0.33521 -1.90221 -0.08503 |
下面我们来看看上面的这个例子里每句话说的是什么意思。
%nprocshared=28 (使用的CPU个数,这个是根据你用的电脑决定的)
%mem=100GB (使用的内存值,这也是根据你用的电脑决定的)
%chk=TNT.chk (作成的checkpoint file即输出文件的名字)
# opt freq b3lyp/6-31g(d,p) ( route section从这一行起开始,这一串字符声明了你的计算条件。opt=结构最优化,freq=振动数以b3lyp/6-31g(d,p)为泛函/基组进行计算)
(这里的空行代表 route section的结束)
**** (这一行是comment section你可以输入你将要计算的分子的名字之类的)
(这里的空行代表comment section的结束)
0 1 (代表电荷和自旋多重度。两个数字之间一定要空一格。0 1代表电荷为0,多重度是1)
C -4.59255 0.08304 -0.05852 (下面的一大串数字表示分子中各个原子的XYZ轴的坐标信息,也有的input file是用Z-matrix来表示原子位置的)
C -3.07318 0.17974 -0.06585
C -2.35822 1.43873 -0.06606
N -2.97016 2.74777 -0.05954
O -2.21637 3.82816 -0.06039
O -4.14959 2.90090 -0.05332
C -0.94837 1.40993 -0.07316
C -0.22055 0.22236 -0.07994
N 1.20996 0.24774 -0.08700
O 1.85923 1.39310 -0.08718
O 1.83665 -0.76914 -0.09283
C -0.91178 -0.98707 -0.07972
C -2.31693 -1.04992 -0.07289
N -2.91158 -2.36710 -0.07344
O -2.14448 -3.43785 -0.08011
O -4.09243 -2.53018 -0.06800
H -5.18863 0.99767 -0.05309
H -4.91946 -0.45909 0.85345
H -4.92857 -0.45422 -0.97006
H -0.39416 2.33820 -0.07345
H -0.33521 -1.90221 -0.08503
(这里的空行代表上面的分子结构信息输入完毕)
(如果有需要增加的指令,在这一行输入,具体参照例8)
(这里的空行表示上行的增加指令已经结束)
(这里的空行表示input file到此全部结束) |
这就是一个input file里那些数字字母所要传达的信息。
例2) 以M062X/6-31g(d,p)为泛函/基组的激发态计算(S1构造最优化,振动数计算)
%mem=100GB
%nprocshared=28
%chk=TNT.chk
# opt freq td=(singlets,nstates=10,root=1) b3lyp/6-31g(d,p)
****
0 1
C -4.59255 0.08304 -0.05852
C -3.07318 0.17974 -0.06585
C -2.35822 1.43873 -0.06606
N -2.97016 2.74777 -0.05954
O -2.21637 3.82816 -0.06039
O -4.14959 2.90090 -0.05332
C -0.94837 1.40993 -0.07316
C -0.22055 0.22236 -0.07994
N 1.20996 0.24774 -0.08700
O 1.85923 1.39310 -0.08718
O 1.83665 -0.76914 -0.09283
C -0.91178 -0.98707 -0.07972
C -2.31693 -1.04992 -0.07289
N -2.91158 -2.36710 -0.07344
O -2.14448 -3.43785 -0.08011
O -4.09243 -2.53018 -0.06800
H -5.18863 0.99767 -0.05309
H -4.91946 -0.45909 0.85345
H -4.92857 -0.45422 -0.97006
H -0.39416 2.33820 -0.07345
H -0.33521 -1.90221 -0.08503 |
例3) 以cam-B3LYP/6-31g(d,p) 为泛函/基组的激发态计算(S0构造)
%mem=100GB
%nprocshared=28
%chk=TNT.chk
# sp td=(singlets,nstates=10,root=1) cam-b3lyp/6-31g(d,p) Guess=Read Geom=AllCheckpoint
**** |
这里的“Guess=Read Geom=AllCheckpoint”是个很好用的操作,可以将之前执行的计算结果和构造不做改动直接用到其他计算任务里。具体应用场景见下面的例4。
例4) NTO计算(S1跃迁轨道的可视化)
%mem=100GB
%nprocshared=28
%chk=TNT.chk
# M062X/6-31g(d,p) Geom=AllCheck Guess=(Read,Only) Density=(Check,Transition=1) Pop=(Minimal,NTO,SaveNTO)
0 1 |
这是标准的模板。如果不想覆盖原有的chk file的话,改成下面这样:
%mem=100GB
%nprocshared=28
%oldchk=TNT.chk
%chk=TNT_NTO.chk
# M062X/6-31g(d,p) Geom=AllCheck Guess=(Read,Only) Density=(Check,Transition=1) Pop=(Minimal,NTO,SaveNTO)
0 1 |
这样,计算结果就不会覆盖原有的chk file而是新输出一个chk file。
例5) 以M062X/6-31g(d,p)为泛函/基组的激发态计算(T1结构最优化,振动数计算)
%mem=100GB
%nprocshared=28
%chk=TNT.chk
# opt freq td=(50-50,nstates=10,root=1) b3lyp/6-31g(d,p)
****
0 1
C -4.59255 0.08304 -0.05852
C -3.07318 0.17974 -0.06585
C -2.35822 1.43873 -0.06606
N -2.97016 2.74777 -0.05954
O -2.21637 3.82816 -0.06039
O -4.14959 2.90090 -0.05332
C -0.94837 1.40993 -0.07316
C -0.22055 0.22236 -0.07994
N 1.20996 0.24774 -0.08700
O 1.85923 1.39310 -0.08718
O 1.83665 -0.76914 -0.09283
C -0.91178 -0.98707 -0.07972
C -2.31693 -1.04992 -0.07289
N -2.91158 -2.36710 -0.07344
O -2.14448 -3.43785 -0.08011
O -4.09243 -2.53018 -0.06800
H -5.18863 0.99767 -0.05309
H -4.91946 -0.45909 0.85345
H -4.92857 -0.45422 -0.97006
H -0.39416 2.33820 -0.07345
H -0.33521 -1.90221 -0.08503 |
这不是针对T1态的构造优化,而是分子会趋向于落入能量最低的激发状态。通常情况下,分子会趋向于处于T1态。
例5-2)不用td的T1态构造最优化
%mem=100GB
%nprocshared=28
%chk=TNT.chk
# opt freq b3lyp/6-31g(d,p)
****
0 3
C -4.59255 0.08304 -0.05852
C -3.07318 0.17974 -0.06585
C -2.35822 1.43873 -0.06606
N -2.97016 2.74777 -0.05954
O -2.21637 3.82816 -0.06039
O -4.14959 2.90090 -0.05332
C -0.94837 1.40993 -0.07316
C -0.22055 0.22236 -0.07994
N 1.20996 0.24774 -0.08700
O 1.85923 1.39310 -0.08718
O 1.83665 -0.76914 -0.09283
C -0.91178 -0.98707 -0.07972
C -2.31693 -1.04992 -0.07289
N -2.91158 -2.36710 -0.07344
O -2.14448 -3.43785 -0.08011
O -4.09243 -2.53018 -0.06800
H -5.18863 0.99767 -0.05309
H -4.91946 -0.45909 0.85345
H -4.92857 -0.45422 -0.97006
H -0.39416 2.33820 -0.07345
H -0.33521 -1.90221 -0.08503 |
注意,与S0的结构最优化和振动数计算的模板不同的地方在于粗体字换成了03。
例6) 只优化氢原子的构造(用于XRD结晶构造解析后的构造最优化)
%nprocshared=28
%mem=100GB
%chk=TNT.chk
# opt freq b3lyp/6-31g(d,p)
****
0 1
C -1 -4.59255 0.08304 -0.05852
C -1 -3.07318 0.17974 -0.06585
C -1 -2.35822 1.43873 -0.06606
N -1 -2.97016 2.74777 -0.05954
O -1 -2.21637 3.82816 -0.06039
O -1 -4.14959 2.90090 -0.05332
C -1 -0.94837 1.40993 -0.07316
C -1 -0.22055 0.22236 -0.07994
N -1 1.20996 0.24774 -0.08700
O -1 1.85923 1.39310 -0.08718
O -1 1.83665 -0.76914 -0.09283
C -1 -0.91178 -0.98707 -0.07972
C -1 -2.31693 -1.04992 -0.07289
N -1 -2.91158 -2.36710 -0.07344
O -1 -2.14448 -3.43785 -0.08011
O -1 -4.09243 -2.53018 -0.06800
H 0 -5.18863 0.99767 -0.05309
H 0 -4.91946 -0.45909 0.85345
H 0 -4.92857 -0.45422 -0.97006
H 0 -0.39416 2.33820 -0.07345
H 0 -0.33521 -1.90221 -0.08503 |
原子右侧的数字-1声明固定的意思,0声明可以移动的意思。即上面的一大串程序表明计算机在对分子作结构最优化时只被允许移动H原子。
如果需要计算的分子太庞大以至于画出来很费时间,我会用下面的Python代码准备gjf file。
filename_input = “./A.gjf”
filename_output = “./output.gjf”
start_line_num = 9
output_text = “”
with open(filename_input, “r”) as f:
lines = f.readlines()
for line_num, line in enumerate(lines):
line = line.strip() # remove break line
if line_num+1 < start_line_num: # line_num starts from 0
output_text += “{}\n”.format(line) # add break line at the end
else:
line_array = [e for e in line.split(” “) if e != “”] # split input line by space and make an array
if len(line_array) > 1:
if line_array[0] == “H”:
line_array.insert(1, str(0)) # add “0” in case of “H”
else:
line_array.insert(1, str(-1)) # otherwise, add “1”
line_output = “\t”.join(line_array)
output_text += “{}\n”.format(line_output) # add break line at the end
# print(“=== output text ====”)
# print(output_text)
with open(filename_output, “w”) as f_out:
f_out.write(output_text)
f = open(‘output.gjf’, ‘a’, encoding=’utf-8′, newline=’\n’)
f.write(‘\n’)
f.write(‘\n’)
f.close() |
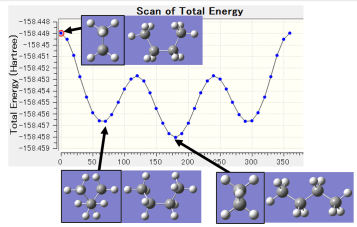
例7) 丁烷的旋转﹑势能面的计算
%chk=butane_scan2
%mem=100GB
%nproc=36
#p opt=modredundant b3lyp/6-31g(d) pop=full
memo
0 1
C -1.81226530 -0.33960474 -0.18951040
H -2.75438476 -0.79975943 -0.40297839
H -1.10701678 -1.08952983 0.10227189
C -1.29892308 0.38635154 -1.44691537
H -2.00326075 0.26246181 -2.24281724
H -0.35589216 -0.02730756 -1.73756778
C -1.12912084 1.88597588 -1.14049402
H -0.42506352 2.00984515 -0.34434097
H -0.77209312 2.39031163 -2.01403661
H -2.07222653 2.29971720 -0.85020142
C -1.98469104 0.67604887 0.95518044
H -2.68960723 1.42622055 0.66322903
H -2.34178564 0.17172469 1.82870237
H -1.04248246 1.13588249 1.16894671
D 11 1 4 7 S 36 10.000000
|
例8) 根据原子指定基函数时
%nprocshared=8
%mem=8GB
%chk=tellurophene.chk
# opt freq M062X/genecp
****
0 1
C 1.30655 0.82344 0.00492
C 2.77391 0.99154 0.00168
N 3.31017 2.23958 -0.00402
C 4.65665 2.40580 -0.00701
N 5.47368 1.32308 -0.00430
C 4.94379 0.07449 0.00139
N 3.59724 -0.09075 0.00438
C 0.69855 -0.36588 0.01053
C -0.68897 -0.37931 0.01309
C -1.31990 0.79801 0.00976
C -2.79024 0.93767 0.01194
N -3.59244 -0.16036 0.01764
C -4.94194 -0.02122 0.01962
N -5.49593 1.21687 0.01592
C -4.70003 2.31522 0.01024
N -3.35058 2.17509 0.00826
Te -0.02269 2.46360 0.00106
H 5.07835 3.40146 -0.01154
H 5.59443 -0.78913 0.00356
H 1.25457 -1.29508 0.01308
H -1.22688 -1.31910 0.01765
H -5.57573 -0.89727 0.02416
H -5.14094 3.30252 0.00729
Te 0
lanl2dz
****
H C N 0
6-31G**
****
Te 0
lanl2dz
|
请注意gen/genecp是关键词,很多人在做含有重原子的计算时经常用吧。最后的黑体字的8行的作用是制定了不同原子的基函数还有Effective Core Potentials (ECP)。
结束语
读者们对每个input file的每段话的大概含义都知道了吧!知道DFT计算是计算什么的非常重要。没必要等到吃透量子化学后才开始用软件计算,即使你对量子化学仍是半瓶水水平,我仍然鼓励你去积极使用软件计算。
希望本篇内容能帮助一些想学习量子计算的同学们找到一些门道。笔者自身水平有限,同时欢迎各位读者的批评和指正,欢迎大家一起交流讨论!
在这里欢迎大家踊跃将自己知道的好的Input File模板分享出来供大家交流学习。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.




















No comments yet.