
近日,国际化学期刊Science Advances(Sci. Adv.)在线发表题为 “A general photoinduced manganese- catalyzed platform for the sequential difunctionalization of [1.1.1]propellane” 的研究论文。该工作由杭州师范大学章鹏飞和浙江大学王华敏团队合作完成。
正文
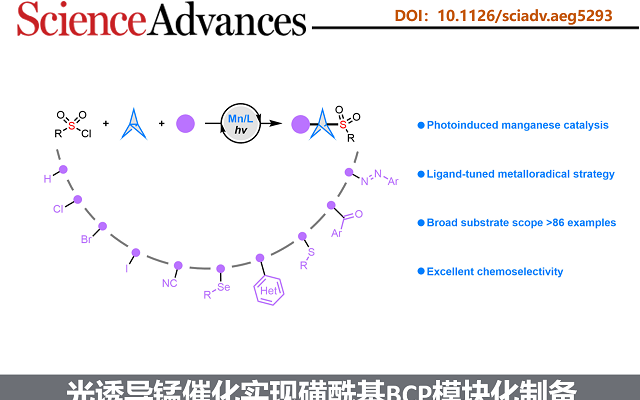
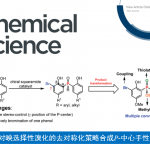
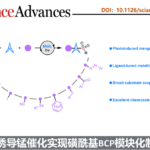
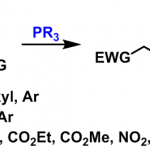
双环[1.1.1]戊烷(BCP)作为苯环的三维生物电子等排体,具有重要的药物化学价值;而磺酰基则是诸多已上市药物中的核心药效基团。将这两类优势结构单元有机结合,有望显著提高分子的代谢稳定性,并增强其整体成药潜力。然而,目前合成含磺酰基BCP衍生物的方法仍存在明显局限:单取代产物的制备通常依赖繁琐的氧化步骤,且所用氧化剂安全隐患较大;双取代衍生物的合成则高度依赖于特定双官能化试剂,限制了分子骨架的多样性;此外,传统策略多采用贵金属光催化剂,且单电子转移(SET)机制容易引发不良副反应。受配体促进光诱导锰催化等前期研究(Fadeyi et al. Angew. Chem. Int. Ed. 2017, 56, 15309; Wang et al., Science 2018, 362, 225; Xie et al., Nat. Synth. 2022, 1, 475; Zhang et al. Green Chem. 2023, 25, 4122)的启发,团队通过调控配体以优化锰中心的电子密度和空间环境,成功构建了高效的锰催化体系,实现了[1.1.1]螺桨烷的可控双官能化。该催化平台兼容氢、氯、溴、碘、氰基、芳硫/硒基、杂芳基、芳酰基、偶氮等多种官能团,并适用于药物分子的后期衍生修饰,为多样化磺酰基BCP生物电子等排体的模块化合成提供了一种实用且高效的方法。(图一)。
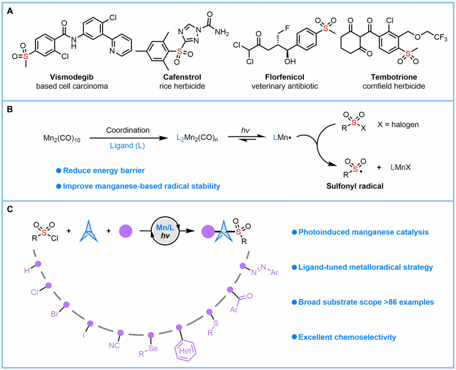
图一:当前的研究现状与反应的设计(图片来源Sci. Adv.)
该研究进一步对反应机理开展了系统阐明。首先,向体系中分别加入TEMPO、DPE、BHT等自由基捕获剂后,反应受到显著抑制,同时高分辨质谱可检测到相应的自由基加成产物,直接证实该转化遵循自由基反应途径(图二A)。其次,采用DTBP、BPO、AIBN等热型自由基引发剂替代标准光催化条件,均无法有效驱动反应进行,表明该转化必须依托光激发锰催化体系,单纯热引发自由基链难以实现目标产物生成(图二B)。光开关间歇实验结果表明,体系中不存在长程自由基链式循环,但反应过程中瞬态锰自由基的二聚过程仍可能发生(图二C)。紫外-可见吸收光谱测试证实,Mn2(CO)10与膦配体L4可原位生成双核金属配合物,其吸收光谱发生明显红移,能够更高效地捕获可见光,从而显著提升锰自由基的生成效率(图二D)。动力学对照实验进一步直观佐证配体具有显著的速率加速作用,移除配体后产物收率大幅降低(图二E)。基于上述实验结果及DFT计算,团队提出了可能的催化反应机理(图二F):在可见光照射下,原位形成的双核锰配合物发生Mn–Mn键均裂,生成锰中心金属自由基A;该自由基通过卤素原子转移(XAT)过程攫取磺酰氯分子中的氯原子,产生磺酰自由基B和金属配合物C。随后,磺酰自由基B对[1.1.1]螺桨烷的张力碳–碳键进行加成,生成BCP自由基中间体D;D进一步进攻含氮杂环,得到中间体E;E与金属配合物C作用,最终生成目标产物,并再生锰自由基A,完成催化循环。
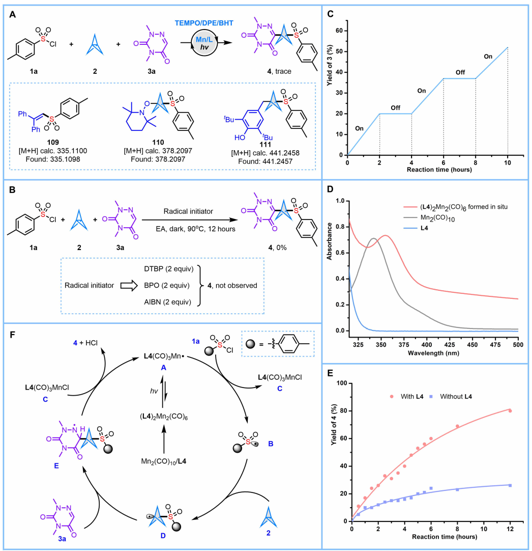
图二:机理探究(图片来源Sci. Adv.)
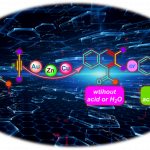
该研究构建了一种可见光驱动的锰催化反应体系,依托配体促进的卤素原子转移(XAT)机制,实现了[1.1.1]螺桨烷的高效连续双官能化转化,从而能够模块化构建结构多样的磺酰基双环[1.1.1]戊烷(BCP)衍生物。该策略不仅突破了传统锰光催化体系的应用瓶颈,也为基于张力释放的自由基多组分反应提供了创新的设计范式。
文章链接:https://www.science.org/doi/10.1126/sciadv.aeg5293














No comments yet.