
作者:孙建松小组
导读
近日,江西师范大学刘慧博士、江南大学孙建松教授和复旦大学李明清研究员合作团队在《JACS Au》上发表了题为[ First Total Synthesis and Bioactivity Study of Dumortierinoside A and Concinnoside D: Two Representative D/E-Ring Simultaneously Functionalized Oleanane-Type Triterpene Saponins]的论文。孙建松教授团队在活性天然皂苷的化学合成领域取得重要突破,成功实现两种结构复杂的天然皂苷–Dumortierinoside A 和 Concinnoside D 的首次化学合成,并深入验证了它们显著的抗癌活性。江西师范大学硕士生高歌和刘慧博士为本文第一作者,刘慧博士、孙建松教授和复旦大学李明清研究员为本文共同通讯作者。
“First Total Synthesis and Bioactivity Study of Dumortierinoside A and Concinnoside D: Two Representative D/E-Ring Simultaneously Functionalized Oleanane-Type Triterpene Saponins”
Ge Gao, Hui Liu*, Xing Zhang, De-Yong Liu, Jin-Xi Liao, Ming-Qing Li*, and Jian-Song Sun*
JACS Au 2025. Doi: 10.1021/jacsau.5c00574
正文
三萜皂苷作为二级代谢产物广泛存在于植物王国中,中药更是三萜皂苷的宝库,如人参、黄芪等传统名贵中药中的三萜皂苷的含量很高,且在药效发挥中扮演着决定性的作用。随着现代分离鉴定手段以及分子生物学技术的快速发展,天然存在着的三萜皂苷优异的生物活性和良好的药用前景逐渐被人们认识。主要表现在抗肿瘤、免疫调节、抗病毒以及降低血糖浓度等方面。作为齐墩果烷型三萜皂苷的重要组成部分,D/E官能团化的齐墩果烷型三萜皂苷因其优异的生物活性越来越引起人们的关注。
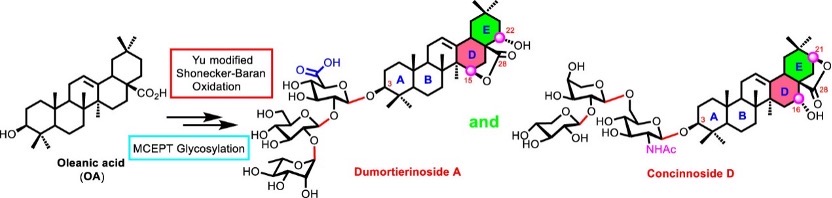
近日,江西师范大学孙建松教授团队在活性天然皂苷的化学合成领域取得重要突破,成功实现两种结构复杂的天然皂苷–Dumortierinoside A 和 Concinnoside D 的首次化学合成,并深入验证了它们显著的抗癌活性。这两种天然皂苷均属于 D/E 同时官能团化的齐墩果烷型三萜皂苷,化学结构复杂。通过传统植化分离,从天然来源提取这两种皂苷面临诸多困难:不仅自然含量低,而且提取过程繁琐、成本高昂,极大地限制了对其药理活性的深入研究与开发应用。团队巧妙运用俞飚院士团队确立改进版 Schonecker−Baran 氧化反应作为关键反应,成功实现了其苷元的高效合成。在此基础上,结合课题组自主研发的新型糖苷化方法–MCEPT 糖苷化方法(J. Am. Chem. Soc. 2023, 145, 3682−3695)以及 DMNPA 保护基(Org. Lett. 2019, 21, 8049−8052; Org. Lett. 2019, 21, 8713−8717),高效实现了其糖链的引入。最终团队分别以最长线性步骤 24 步、25 步,总产率 2.1%和 1.6%成功完成了 Dumortierinoside A 和 Concinnoside D 的首次全合成。为进一步探究这两种合成皂苷的生物活性,孙建松教授团队与复旦大学李明清课题组展开紧密合作,对合成所得的皂苷化合物进行了全面且深入的抗肿瘤活性评价。实验结果表明,这两种皂苷展现出显著的抗癌活性,为后续的药物研发提供了坚实的理论基础和实验依据。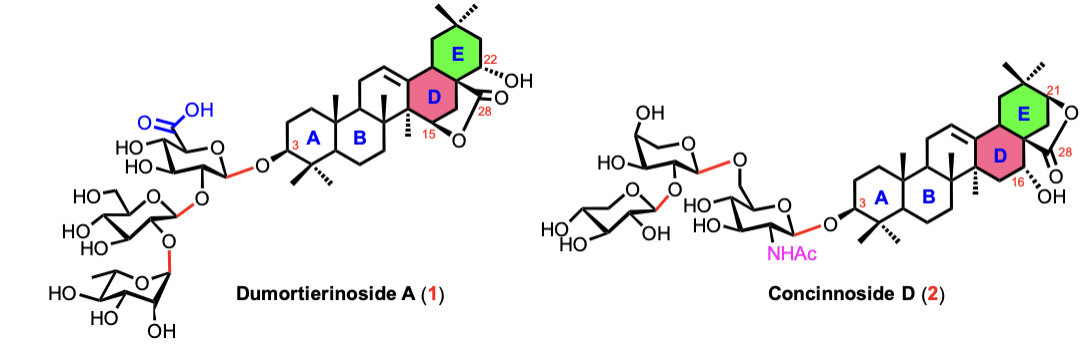
Figure 1. The chemical structures of dumortierinoside A and concinnoside D
作者首先以商业可得的齐墩果酸(Oleanolic acid)为原料,根据文献报道的方法衍生得醛3,之后经连续两步改进版的Schonecker−Baran氧化实现了C22和C16位的C-H活化得化合物6,随后再经关键的4步氧化(Pinnick 氧化、Ley氧化、NaHMDS/O₂/P (OEt)₃、Ley氧化)得到了C15位也被氧化的二酮化合物9,化合物9在SmI2的条件下利用28位甲酯的导向作用选择性将C16还原脱氧得化合物10。化合物10中的15位羰基再经DIBAL-H的立体选择性还原成羟基并随即内酯化得化合物12,同时伴随着内酯进一步被还原的化合物11,幸运的是,化合物11可以在Ley氧化的条件下转化成化合物12。12再进行一步脱保护即可得Dumortierinoside A的甙元13。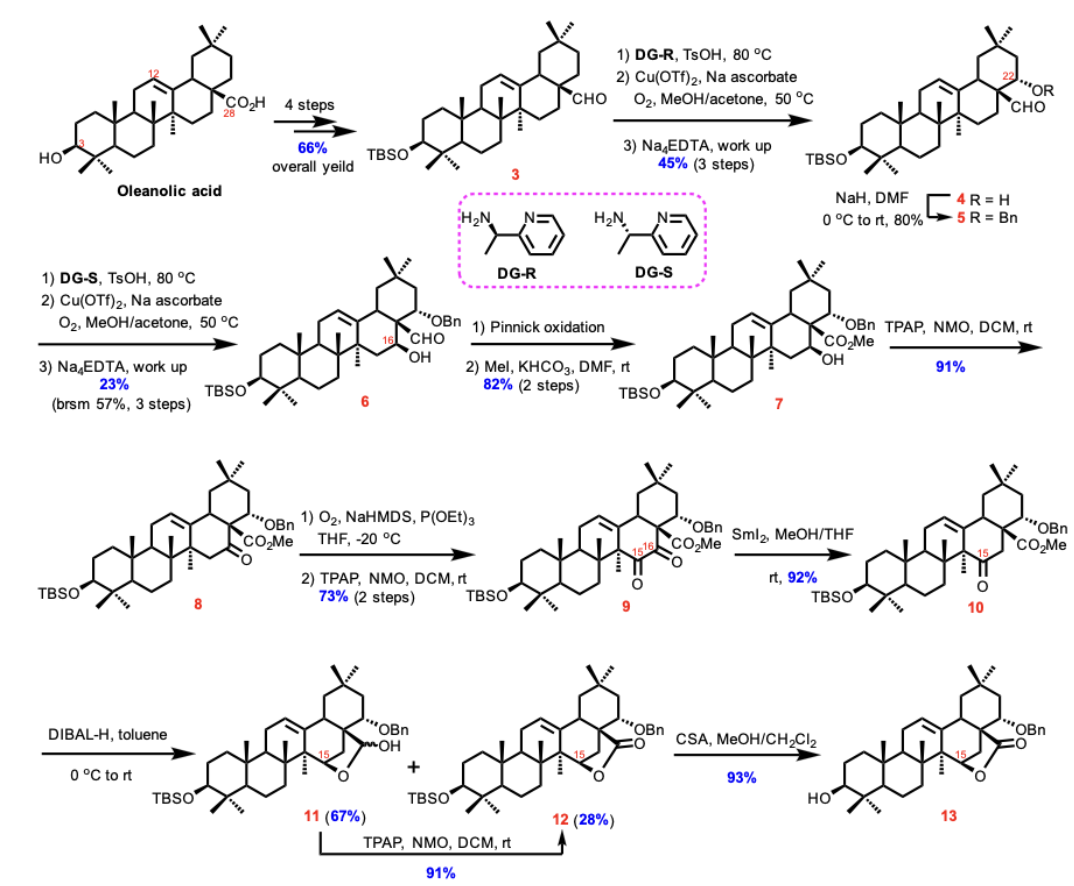
Scheme 1. Synthesis of dumortierigenin aglycone 13.
在这条路线中C16位C-H活化的产率偏低导致总体的效率偏低,所以作者又探索了一条高效制备Dumortierinoside A的甙元的路线,即先利用改进版的Schonecker−Baran氧化对C16进行C-H活化,再消除得化合物15,再利用Schonecker−Baran氧化即可高区域高立体选择性的得化合物16,之后经关键的Vo(acac)2/TBHP立体选择性环氧化反应和LAH区域选择性开环氧得三醇化合物19,之后伯羟基经TEMPO氧化,15位羟基经Misunobu反应进行手性翻转之后发生分子内的内酯化,脱除硅基保护基得到22位羟基未保护的甙元21,由于在研究中作者发现22位羟基的位阻很大,后续糖苷化可以选择性的在C3位羟基上发生。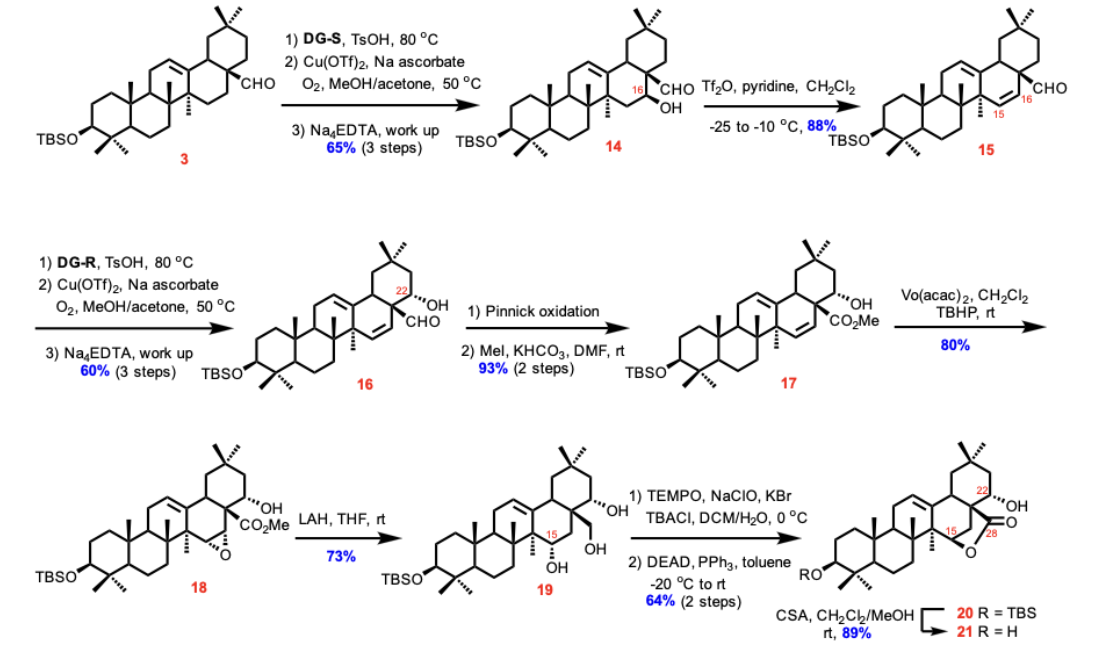
Scheme 2. Synthesis of dumortierigenin aglycone 21.
作者利用课题组自主研发的MCEPT糖苷化方法和DMNPA保护基结合线性合成策略完成了Dumortierinoside A(1)的高效高立体选择性合成。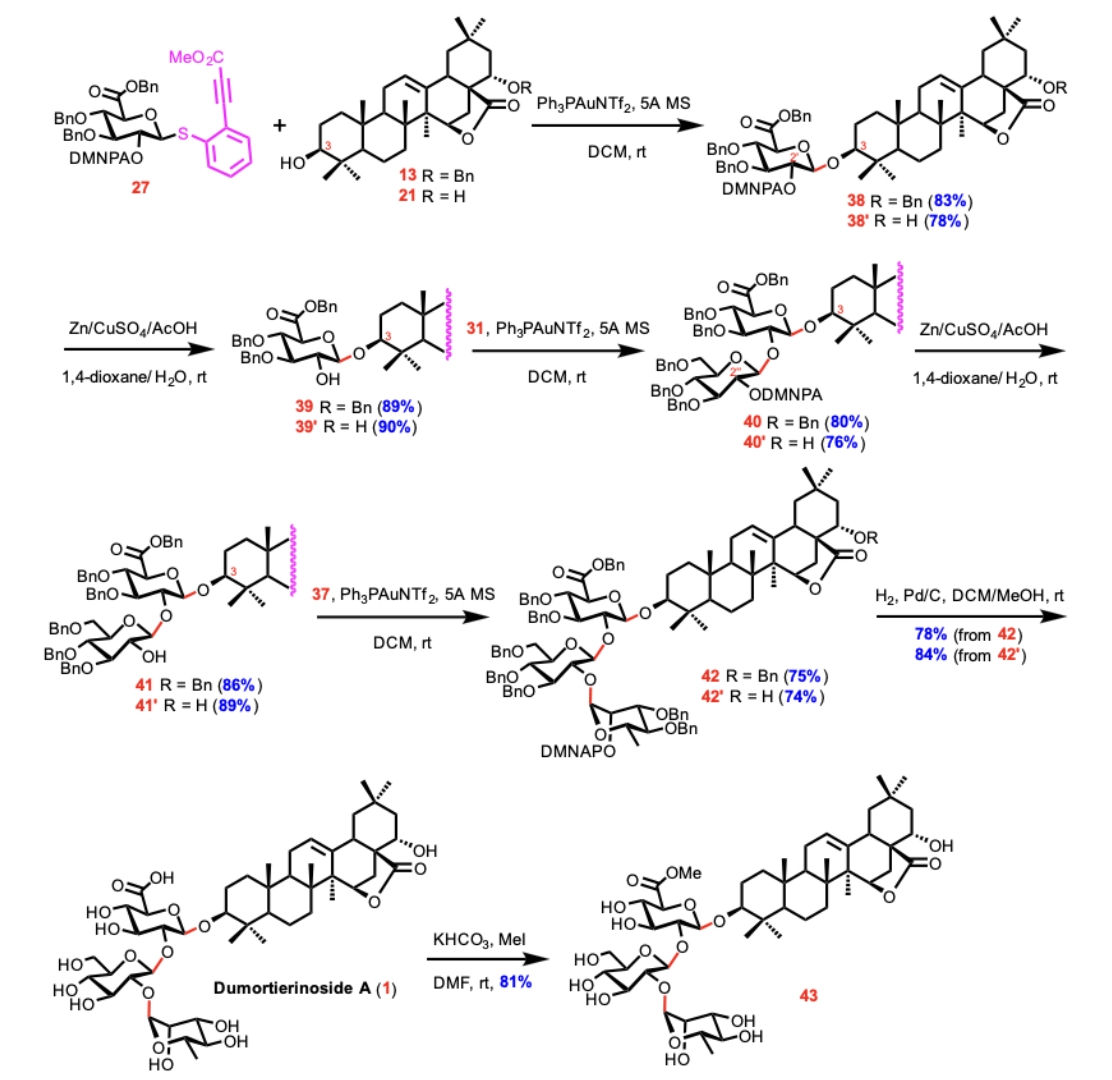
Scheme 3. Completion of the synthesis of dumortierinoside A (1) and the corresponding methyl ester 43.
对于Concinnoside D的甙元金合欢内酯的合成,作者同样是以便宜易得的齐墩果酸为原料,参考俞飚院士报道的工作,经17步转化得三醇化合物44,之后再Anelli氧化的条件下选择性的将伯羟基进行氧化,接着C21位羟基在Misunobu反应的条件下构型翻转紧接着发生内酯化得化合物45,化合物45脱除3位的硅基保护基得Concinnoside D的甙元金合欢内酯46。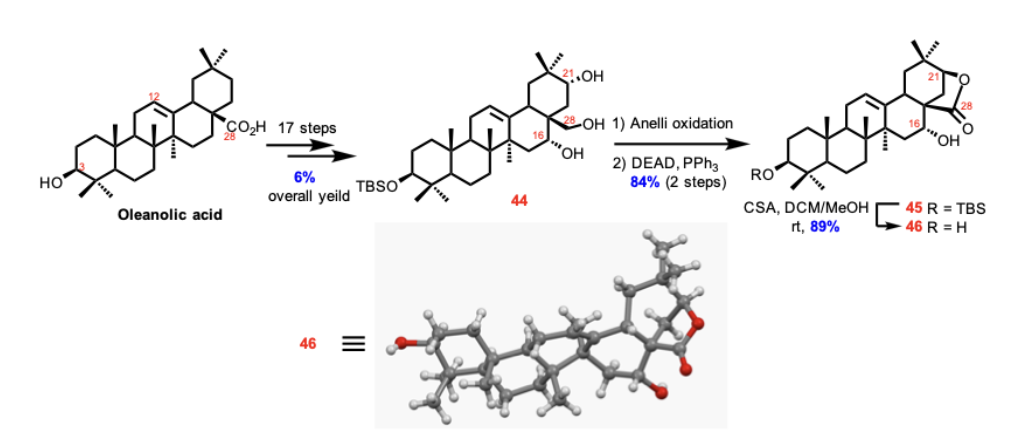
Scheme 5. Synthesis of acacic acid lactone 46.
为了使得合成更为高效,对于糖链的引入,作者采用的是汇聚式的合成策略,利用MCEPT糖苷化方法和潜在活化的糖苷化策略,作者高效制备出合成Concinnoside D所需的三糖给体66,之后三糖MCEPT给体66和金合欢内酯46在PPh3AuNTf2的条件下高效糖苷化,最后脱除保护基得Concinnoside D。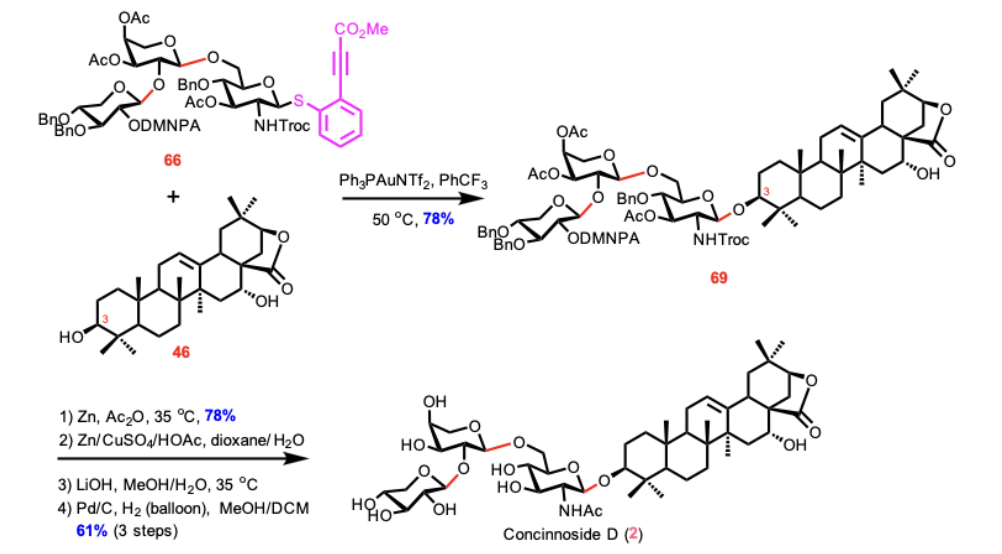
Scheme 8. Completion of the synthesis of concinnoside D (2).
为进一步探究这两种合成皂苷的生物活性,作者与复旦大学李明清课题组展开紧密合作,对合成所得的皂苷化合物进行了抗肿瘤活性评价:采用标准 MTT 法检测其对多种肿瘤细胞的抑制作用:对乳腺癌细胞 MDA-MB-231:Concinnoside D 表现出显著的抑制效果,24 小时和 48 小时的 IC50 值分别为 22.36 μmol 和 22.3 μmol,优于阳性对照顺铂(71.7 μmol 和 29.3 μmol)。对人宫颈癌细胞 HeLa:Dumortierinoside A 展现出明显的细胞毒性,48 小时 IC50 值为 11.14 μmol,与顺铂(5.779 μmol)相当。对人子宫内膜癌细胞 RL95-2:两种皂苷均表现出较强的抑制作用,IC50 值与顺铂相近。
Table 1. Cytotoxic effect of saponins 1 and 2 against tumor cell lines.a
|
Tumor cell linesb |
IC50 (μmol, 24 h)c | IC50 (μmol, 48 h)d | ||||
| 1 | 2 | cisplatin | 1 | 2 | cisplatin | |
| MDA-MB-231 | 453.7 | 22.36 | 71.7 | 152 | 22.3 | 29.3 |
| HeLa | 95.6 | 226.7 | 89.18 | 11.14 | 52.57 | 5.779 |
| RL95-2 | 48.51 | 58.24 | 46.39 | 14.46 | 39.8 | 11.08 |
aThe standard MTT assay was used (MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). bMDA-MB-231 = breast cancer cell line, Hela = human cervical cancer cell line, RL95-2 = human endometrial carcinoma cell line. cDetected at 24 h after treatment with 1, 2, and cisplatin. dDetected at 48 h after treatment with 1, 2, and cisplatin.
小结
孙建松教授团队在活性天然皂苷的化学合成领域取得重要突破,成功实现两种结构复杂的天然皂苷–Dumortierinoside A 和 Concinnoside D 的首次化学合成,并深入验证了它们显著的抗癌活性。其中巧妙运用俞飚院士团队确立改进版 Schonecker−Baran 氧化反应作为关键反应,成功实现了其苷元的高效合成。在此基础上,结合课题组自主研发的新型MCEPT 糖苷化方法以及 DMNPA 保护基高效实现了其糖链的引入。最终团队分别以最长线性步骤 24 步、25 步,总产率 2.1%和 1.6%成功完成了 Dumortierinoside A 和 Concinnoside D 的首次全合成。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
















No comments yet.