
作者:石油醚
导读:
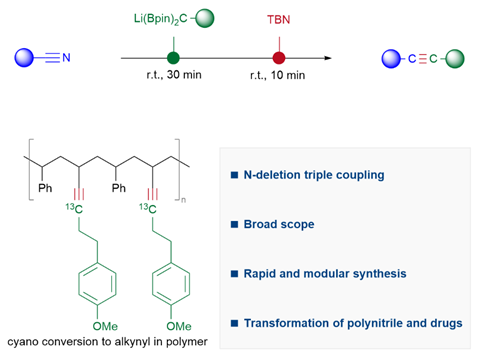
近日,中国科学院兰州化物所刘超(现任职于南京大学)团队开发了一种从腈类化合物出发,利用TBN作为删氮试剂,通过与锂化烷基偕二硼反应合成炔烃的方法,成功地将三键中的氮原子置换为碳原子。在短时间内,包括芳香族和脂肪族在内的腈类化合物可以快速转化为各种内炔和端炔,使得氰基可以等价成炔基标记在功能团转化中。通过这种方法,可以将多氰基化合物快速高效地转化成相应的多炔基化合物。对于氰基的α位和偕二硼中含有大位阻的底物使用这种方法也可以转化成大位阻的炔烃。这一研究成果近期发表在Chem杂志上,在读博士生徐良轩为文章第一作者。该工作受到国家自然科学基金(22022113)的支持。
“Atom Swap in Triple Bonds via Nitrogen Deletion Coupling with gem–Diborylalkanes.
Liangxuan Xu, Du Chen, Peng Zhang*, Chungu Xia*, Chao Liu*
Chem, 2024. Doi: 10.1016/j.chempr.2024.08.001”
正文:
三键是最有价值的官能团之一。三键的不饱和性为进一步转化生成各种分子提供了广大的空间。其线性分子结构和特殊的电子结构使得三键在医药和功能材料中也具有重要价值。炔基和氰基是最重要和最常见的三键官能团,在许多生物活性化合物、药物、农用化学品和众多合成转化中都发挥了关键作用。此外,腈和炔烃还表现出生物异构性,这意味着在药物分子中用炔烃取代腈可以产生新的生物活性特性。例如,用炔烃分子取代一种抗MRSA药物中的氰基,对MRSA菌株的药效显著提高了67倍,同时在小鼠体内保持了令人满意的衰减半衰期。同样,将一种抗艰难梭菌药物的氰基替换为炔基后,其药效提高了16倍以上(图1A)。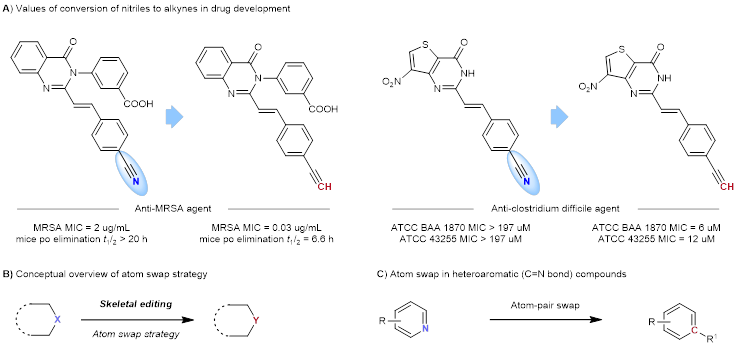
图1. 研究背景
在镍催化的Sonogashira型偶联反应中,氰基作为卤素基团等价物,其中的“CN”被完全取代,从而实现了腈到炔的转化。但C-CN键的惰性限制了此类反应范围。最近,通过原子交换策略进行骨架编辑的概念在合成化学中受到越来越多的关注(图1B)。这种策略的魅力在于可以方便地交换那些具有挑战性的不饱和化学键中的原子。例如,通过N/C交换对芳香族N-杂环(C=N键)进行后期骨架修饰,就可以编辑芳香族化合物(图1C)。氰基和炔基的区别在于“N”原子和“C”原子。基于相应的背景,作者设想能否实现三键中N原子与C原子的互换。如果成功,就可以实现从腈到炔的直接转化。基于文献调研,三键中的原子互换还很少有人探索过。虽然腈的复分解反应已经证明可以将腈转化为炔,但利用钼、钨络合物生成炔烃混合物的腈的复分解反应却存在缺点。
中国科学院兰州化物所刘超(现任职于南京大学)团队长期致力于有机硼化学研究,并取得了一系列研究进展(J. Am. Chem. Soc. 2017,139, 5257-5264; Angew. Chem. Int. Ed. 2018, 57, 10318-10322; Angew. Chem. Int. Ed. 2019, 58, 15813-15818; Angew. Chem. Int. Ed. 2020, 59, 2745-2749; CCS Chem. 2021, 3, 1718-1728; Angew. Chem. Int. Ed. 2024, 63, e202315227)。近年来,该团队围绕有机硼化学发展了一系列新型试剂实现了官能团快速转换反应(Angew. Chem. Int. Ed. 2018, 57, 5501-5505; Nat. Commun. 2020, 11, 3113; Angew. Chem. Int. Ed. 2023,62, e202215168; Nat. Synth. 2023, 2, 413-422)。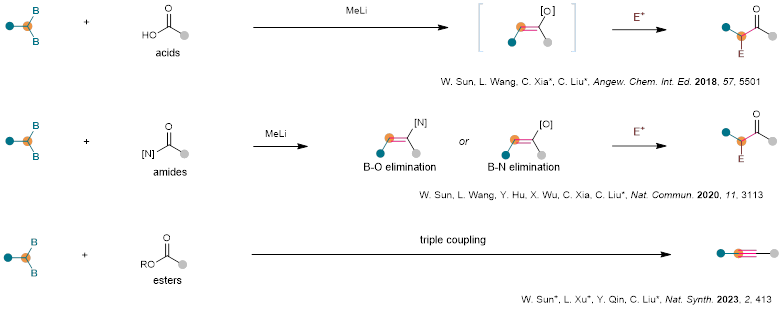
近来,中国科学院兰州化学物理研究所刘超/夏春谷/张鹏团队受到偕二硼化合物与羧酸酯合成炔烃工作的启发,尝试以偕二硼化合物与腈反应经过亲核加成得到的烯酰胺中间体,再经过消除,实现腈到炔烃的直接转化,实现碳氮三键和碳碳三键中的原子交换的过程。在已报道的酯与偕二硼反应实现炔烃的合成中,锂化偕二硼对羰基的加成得到的中间体经历B-O消除得到炔烃。而在腈与偕二硼的反应中,反应历程和酯与偕二硼的反应历程很相似,不同的是此过程需要经历N-B消除。该策略的优点在于:1)反应的中间体Int-I是一种全新的配合物,该中间体可以进一步消除生成炔烃;2)反应体系简单,底物使用范围广。当然,该策略同时也存在着很大的挑战:1)C-N键能很高,导致C-N键的断裂比较困难;2)寻到合适的C-N键的活化试剂比较困难(图2A)。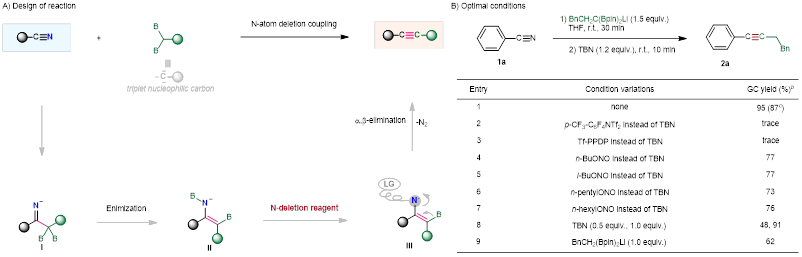
图2. 反应设计及条件优化
根据反应的设计,作者进行了尝试,并成功得到了产物炔烃。在此基础上进行了一系列的优化,得到了最优的反应条件(图2B)。接下来,作者对反应底物的普适性进行了考察(图3)。底物具有很好的官能团兼容性,并且能够快速高效地生成炔烃。该反应重点解决脂肪族炔烃以及炔基两端含有大位阻的难合成的炔烃。同时,手性α-取代腈在不发生消旋的情况下也转化为相应的手性炔丙基化合物。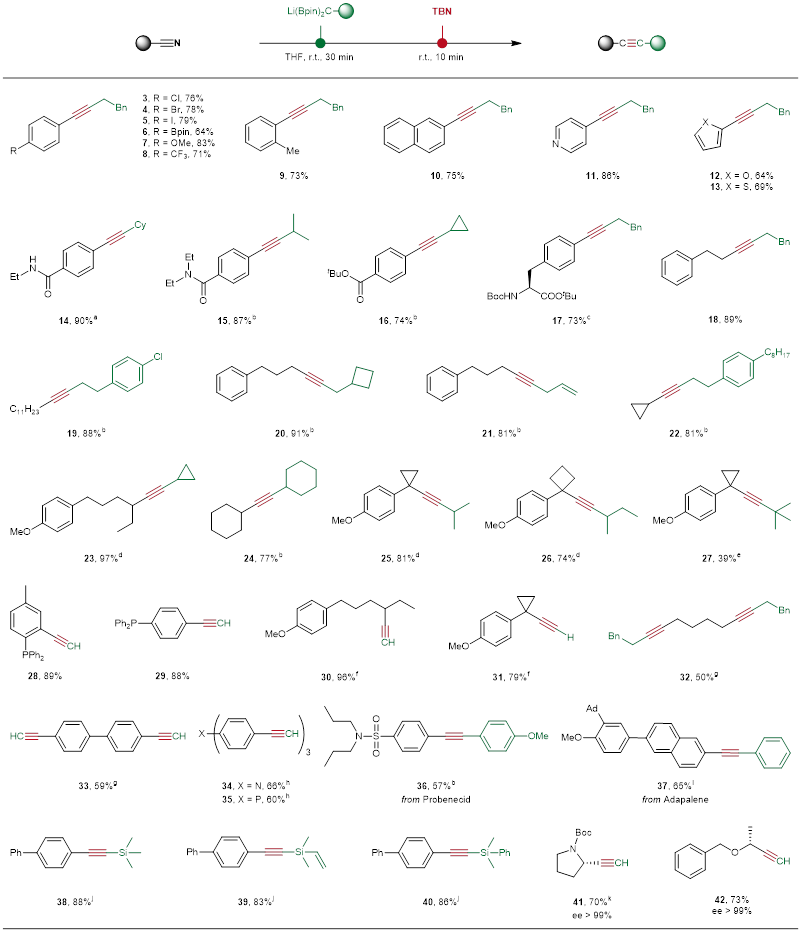
图3. 底物普适性考察
许多天然产物和药物都含有氰基官能团。通过这种三键中原子交换策略直接对含氰基药物分子进行后期修饰将有利于新药开发。在此,作者选择了四种药物分子,包括西酞普兰、非布司他、维拉帕米和阿那曲唑,它们都以良好的收率成功转化为相应的端炔化合物(43–46,图4)。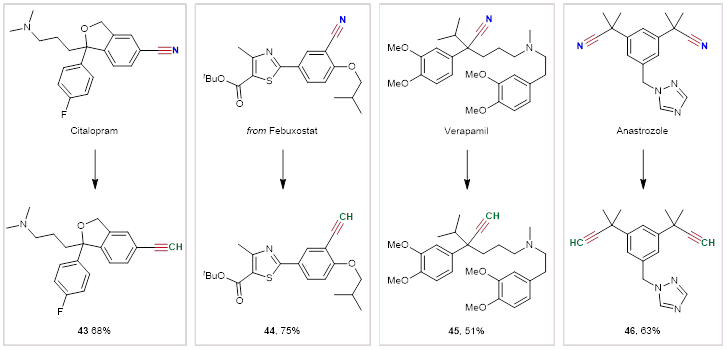
图4. 含氰基的药物分子的修饰
为了进一步证明这种方法的合成价值,作者列出了两种炔烃47和48,以便与已报道的方法进行比较。炔烃47和48已被应用于氧化环化反应。从腈合成这些炔烃(产率较低)需要经过还原成醛、缩合成偕二溴和低温锂化再消除等几个步骤。与此相反,利用所开发的方法成功地从腈出发以高收率一步合成这些炔烃(47, 81%;48, 75%)(图5)。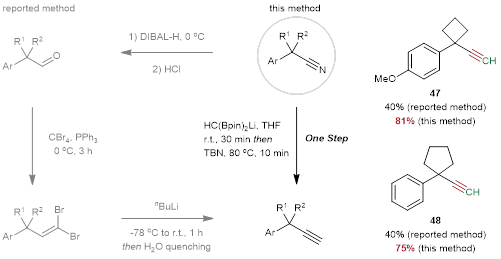
图5. 与传统方法的比较
聚合物腈在人类的生命活动中发挥着重要作用,而且很容易从商业渠道获取。利用这种合成策略,可以使聚苯乙烯–共丙烯腈(CN-49)与对甲氧基苯丙基偕二硼–13C发生反应。通过核磁分析(图6),聚合物中约46%的氰基转化为炔基单元(49)。这一非常有意义的结果将促进聚合物腈的多样化利用。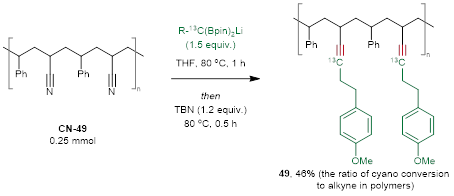
图6. 聚合物腈的三键中的原子交换
为了进一步了解腈与锂化的偕二硼反应生成的中间体的性质,作者进行了原位核磁实验。对同位素富集的13CH(Bpin)2Li与4-甲氧基苯甲腈–13C的反应进行了核磁共振分析(图7)。13CH(Bpin)2Li与1.3当量的4-甲氧基苯甲腈–13C的反应混合物主要显示了归属于α-硼基锂烯酰胺Int-A的信号。通过与非标记的平行反应进行比较,1H NMR谱包含一个对应于位于3.58 ppm处的Hb的峰,它与富集的Ca和Cb(J = 136.0 Hz,2.7 Hz)耦合。13C NMR谱显示,烯酰胺Int-A中Ca和Cb的化学位移分别为180.5 ppm和83.5 ppm。此外,11B NMR谱显示在31.36 ppm、29.58 ppm和24.94 ppm处的宽信号被认为来自原料的C-Bpin、Int-A的C=C-Bpin和Int-A的N-Bpin。与此同时,结合苯甲腈和BnCH2C(Bpin)2Li的反应混合物中得到的单晶分析确定反应中间体为α-硼基锂烯酰胺Int-A。由于螯合效应,硼基位于烯酰胺基团的顺式位置。Ca–Cb键长为1.365 Å,表明具有双键特性。接着TBN对中间体Int-A进行N-重氮化,再通过β-硼消除反应即可立即生成炔烃。通过比较13C标记和非标记反应,从1H NMR和13C NMR中可以明显看出,在加入TBN到反应混合物后,产物1-乙炔基-4-甲氧基苯的生成,显示了所有Int-A到1-乙炔基-4-甲氧基苯的消除。炔基氢的化学位移为3.34 ppm(dd,J = 247.8 Hz,53.0 Hz),Ca的化学位移为84.3 ppm(d,JC-C = 174.0 Hz),Cb的化学位移为77.0 ppm(d,JC-C = 174.0 Hz)。此外,结合11B NMR和标准样品分析,21.3 ppm为tBuOBpin的信号峰。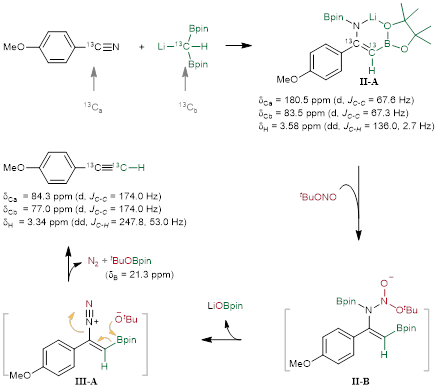
图7. 反应机理研究
综上,该团队开发了一种从腈类化合物出发,利用TBN作为删氮试剂,通过与锂化烷基偕二硼反应合成炔烃的方法,成功地将三键中的氮原子置换为碳原子。在短时间内,包括芳香族和脂肪族在内的腈类化合物可以快速转化为各种内炔和端炔,使得氰基可以等价成炔基标记在功能团转化中。通过这种方法,可以将多氰基化合物快速高效地转化成相应的多炔基化合物。对于氰基的α位和偕二硼中含有大位阻的底物使用这种方法也可以转化成大位阻的炔烃。
(非常感谢刘超教授对Chem-Station的支持)















No comments yet.