
本文作者:杉杉
导读
近日,日本Tohoku大学的Kondo Yoshinori (根東 義則) 与Shigeno Masanori (重野 真徳)研究团队在Org. Lett.中发表论文,报道一种采用偶氮苯作为起始原料,通过邻位亲电硼化与亲核二烷基化关环的两步反应设计方案,进而成功实现各类1,2,3-benzodiazaborole分子的构建。这一全新的合成转化策略具有底物应用范围广泛以及高度的官能团兼容性等优势。同时,作者通过DFT计算表明,带有B-N-N键的五元杂环分子具有中等程度的芳香性。同时,作者进一步对上述反应过程的机理进行深入研究。
Construction of 1,2,3-Benzodiazaborole by Electrophilic Borylation of Azobenzene and Nucleophilic Dialkylative Cyclization
M. Shigeno, M. Imamatsu, Y. Kai, M. Kiriyama, S. Ishida, K. Nozawa-Kumada, Y. Kondo, Org. Lett. 2021, ASAP. doi: 10.1021/acs.orglett.1c03033.
正文
通过等电子 (isoelectronic)与等结构 (isostructural)的B-N键替代芳香化合物中的C=C键是目前药物化学与材料科学相关领域中的重要课题。通过BN/CC电子等排体 (isosterism)的作用,能够在有机分子中引入相应的偶极矩,进而对有机分子的一系列相关性质,例如电子与光致发光特性、理化性质与药代动力学活性以及芳香性与化学反应活性,进行有效调控,并使上述的相关性质与最初的芳香化合物存在显著差异。其中,对于具有六元环结构的苯类似物的相关研究已经取得诸多进展,例如,目前已经成功完成一系列1,2-azaborine,1,3-azaborine以及1,4-azaborine分子及其衍生物的构建[1]。
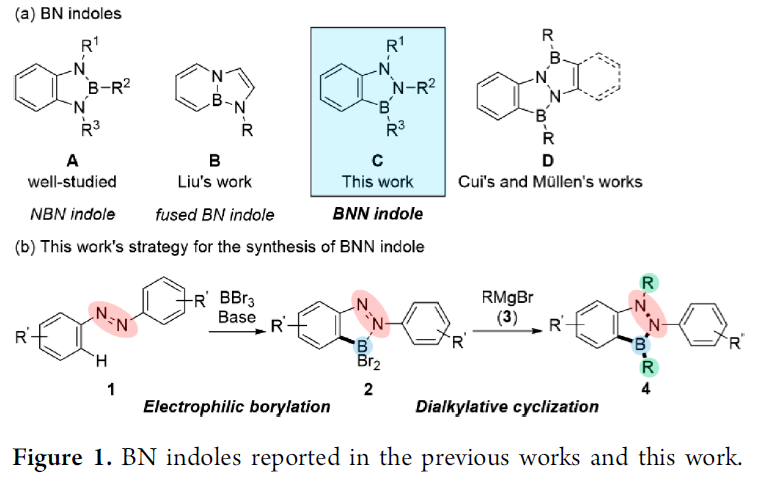
而吲哚骨架是药物化学中的一种十分重要的结构单元或较为关键的药效团 (pharmacophore)。并且,目前已经成功开发出多种能够用于构建一系列官能团化吲哚衍生物的相关反应策略[2]。同时,对于BN类似物以及BN吲哚化学的相关报道,已经成功为当代有机化学的研究开辟出一种全新的方向 (Figure 1a)。其中,1,3,2-benzodiazaborole A (即NBN 吲哚)能够十分容易地通过1,2-二氨基苯与苯硼酸衍生物作为原料进行制备[3],这一反应策略在上世纪中叶首次报道。之后,Liu课题组报道一种稠合BN吲哚分子B (其分子结构中,六元环与五元环通过B-N键进行稠合)的合成策略[4]。近期,Cui与Müllen团队分别报道涉及1,3,2-benzodiazaborole D (即BNN 吲哚) 分子构建的相关策略[5]–[6]。然而,对于简单BNN吲哚核心骨架的构建,目前仍面临诸多挑战。并且,迫切需要设计一种更为高效并通用的反应策略。
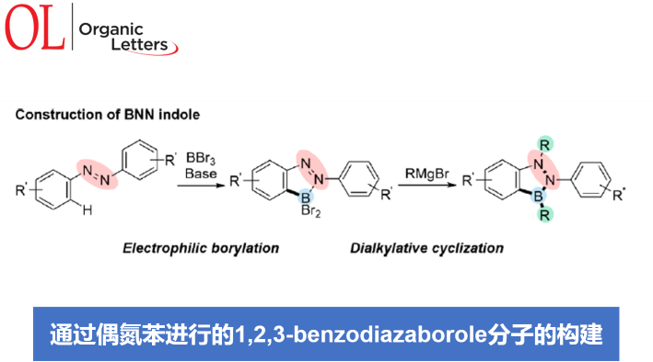
为解决上述问题,本文中,Y. Kondo与M. Shigeno研究团队报道一种采用廉价易得的偶氮苯1作为起始原料,通过采用BBr3进行的邻位亲电硼化以及通过Grignard试剂进行的二烷基化关环的两步反应设计方案,进而顺利实现一系列BNN吲哚分子4 的构建 (Figure 1b)。同时,受到通过邻位导向基团,例如吡啶基、苯胺基团以及N-酰基参与的芳香C-H硼化反应方法学研究的启发[7],作者设想,首先通过偶氮苯1与亲电硼试剂之间的硼化过程,形成具有邻位二溴硼官能团的砌块2。同时,前期已经有文献报道涉及2-(2-二溴硼基苯基)吡啶与金属有机亲核试剂在硼原子位置进行的双重取代过程,并获得具有二烷基硼基团的有机分子[8]。然而,由于通过偶氮基团中氮原子与BBr2结构单元的配位,使砌块2的亲电性能显著增强,进而能够有效地促进偶氮官能团的亲核加成以及后续B-Br键亲核取代过程的进行。这里,本文将对上述的两步反应设计方案的可行性进行深入研究。并且,通过相关研究,进一步设计出一种构建BNN吲哚分子的高效并通用的反应策略。
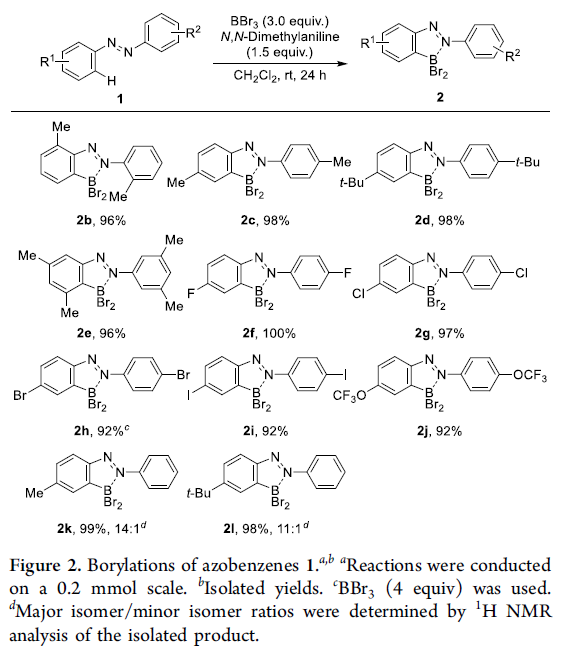
首先,作者采用偶氮苯1a与BBr3作为模型底物,进行亲电硼化反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用N,N-二甲基苯胺作为碱,在二氯甲烷溶剂以及室温条件下进行反应,最终获得98%收率的硼化产物2a。

在上述的标准硼化反应条件下,作者对各类偶氮苯底物1的应用范围进行考察 (Figure 2)。研究表明,苯环不同位置中具有一系列供电子与吸电子基团取代的偶氮苯底物,均能够较好地与上述的标准硼化反应条件兼容,并获得相应的目标产物2b–2j,收率为92-100%。接下来,该小组观察到,对于不对称底物1k与1l参与的硼化过程,则区域选择性地在底物中更为富电子的苯环位置进行。接下来,作者发现,将底物1a的用量扩大至10 mmol时,同样能够获得95%收率的硼化产物2a (Scheme S3)。
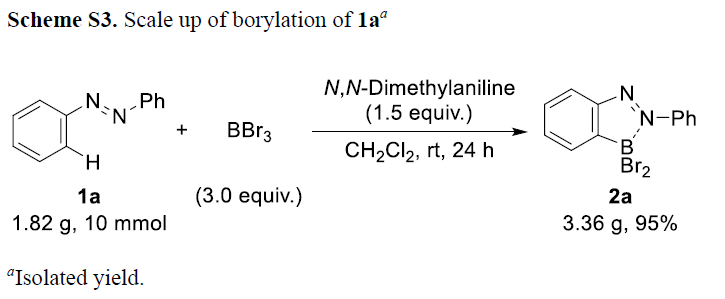
为阐明上述硼化反应的合成实用性,作者进一步对相应硼化产物进行一系列相关的合成转化研究。该团队发现,硼化产物中的BBr2结构单元能够有效地进行一系列不同类型的合成转化过程,主要涉及氧化、水解、频哪醇保护、1,2-苯二胺保护以及采用Zn(C6F5)2试剂进行的二芳基化反应 (Figure S1)。
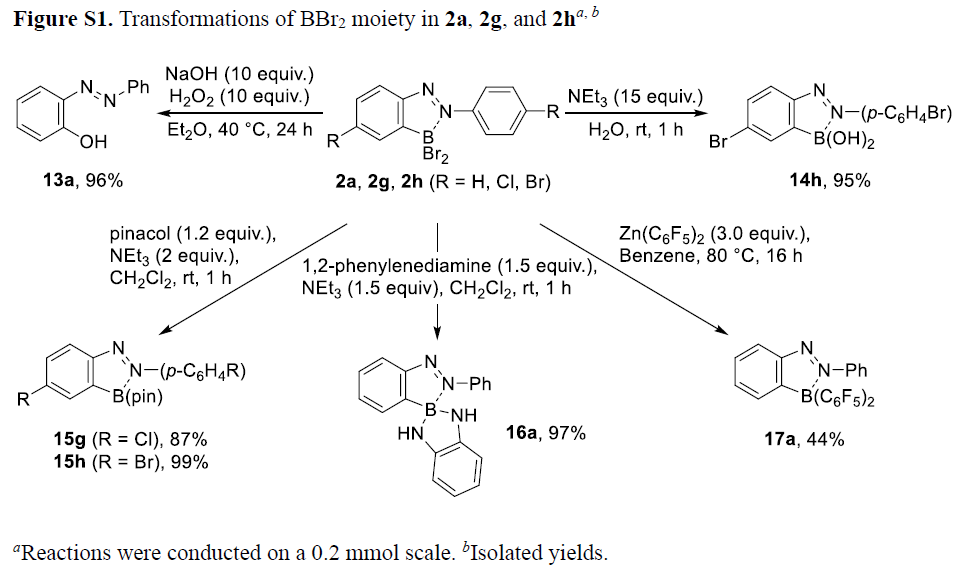
接下来,为进行硼化偶氮苯类化合物分子轨道的相关研究,作者在B3LYP/6-311G(d,p)水平下,进行相应的DFT计算 (Figure 3)。研究表明,与1a (-2.43 eV)相比,通过BBr2结构单元的引入,使2a 分子的LUMO能级 (ELUMO)显著降低至-3.67 eV,并且,2a中的LUMO主要位于偶氮基团的位置。
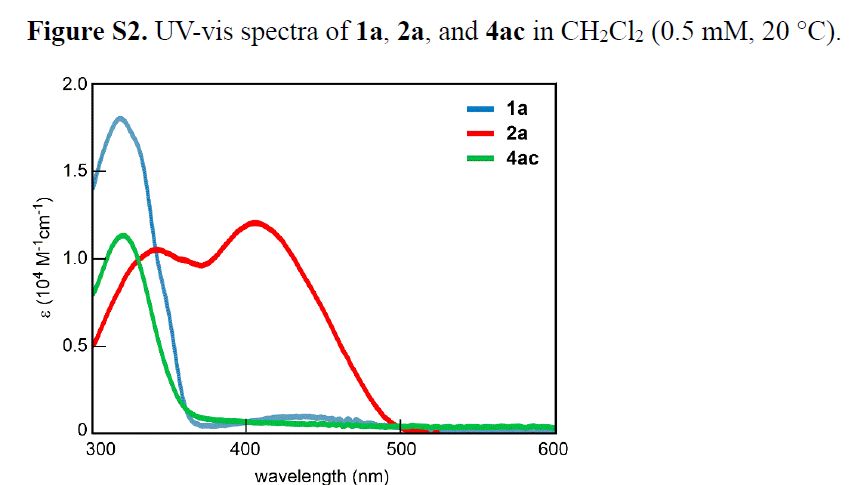
之后,作者发现,2a分子的UV -Vis光谱中,在λmax = 407 nm处出现最大吸收峰,这一吸收峰与偶氮基团的π-π*跃迁相对应。同时,与1a 分子相比 (λmax = 318 nm),2a分子的吸收峰位置出现较为显著的红移 (Figure S2)。
接下来,作者采用2a作为模型底物,进行相应烷基化关环反应条件的优化筛选 (Table 2)。进而确定最佳的烷基化关环反应条件为:采用Grignard试剂MeMgBr,二氯甲烷作为反应溶剂以及反应温度为0 oC,并获得94%收率的BNN吲哚产物4aa。

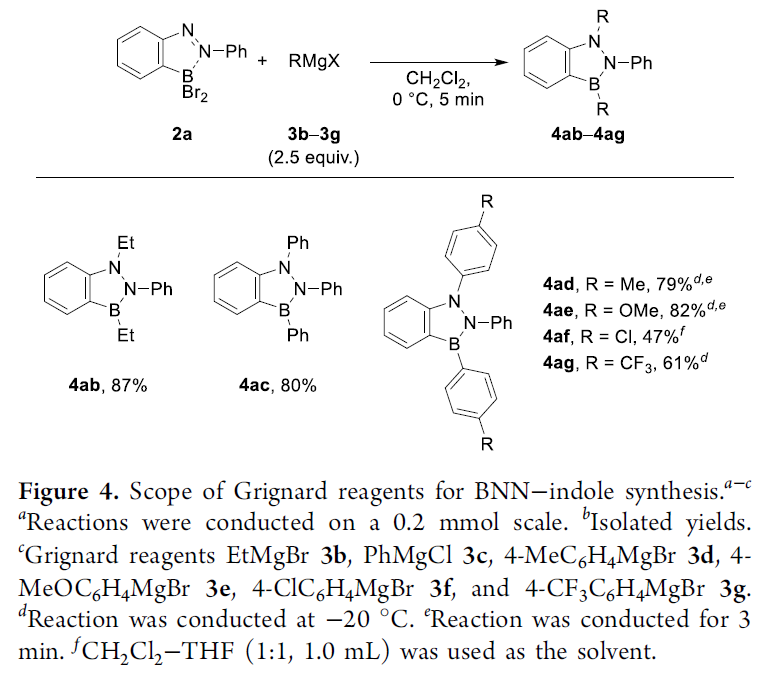
之后,作者对相应Grignard试剂的应用范围进行深入研究 (Figure 4)。实验发现,采用EtMgBr试剂,同样能够获得87%收率的目标产物4ab。同时,作者观察到,选择亲核反应活性较低 (与EtMgBr试剂相比)的试剂,例如AlEt3或ZnEt2,则使目标产物4ab 的收率出现显著降低 (Table S3)。并且,该小组进一步发现,一系列不同具有基团取代的芳基Grignard试剂,均能够顺利地参与上述的烷基化关环反应过程,并获得相应的BNN吲哚产物4ac–4ag,收率为47-82%。
同时,作者同样对硼化偶氮苯底物的应用范围进行进一步考察 (Figure 5)。研究表明,底物中苯环不同位置具有甲基、叔丁基、卤原子以及三氟甲氧基基团取代的硼化偶氮苯底物,均能够顺利地与3a或3c进行反应,并获得相应的BNN吲哚产物,收率为68-94%。

接下来,作者在B3LYP/6-311+G(2d,p)水平下,对4ac分子进行核独立化学位移值 (nucleus-independent chemical shift, NICS) 的相关计算,最终获得NICS(1)zz的数值为-11.8 ppm。进而表明,带有B-N-N键的五元杂环分子,具有中等程度的芳香性 (Figures 5-6)。

为进一步阐明烷基化环化过程的反应机理,作者进行一系列相关的实验研究 (Scheme 1)。该小组观察到,在2a依次采用MeMgBr与PhMgCl进行反应时,能够获得BNN吲哚产物5,其中,甲基进入2a中的氮原子位置,苯基进入2a中的硼原子位置。同时,该小组进一步发现,实验过程中,将上述的两种Grignard试剂3a与3c按相反的顺序进行时,则获得相反区域选择性的BNN吲哚产物6。

同时,作者假设,在两种可能的机理路径 (Scheme 2)。而基于上述的实验研究,作者推测,Path A更为合理。首先,亲核试剂与底物中的偶氮基团通过亲核加成以及后续的分子内环化过程,形成具有 B-N-N键的五元环化合物7。接下来,通过亲核试剂对7 中B-Br键的亲核进攻过程,最终获得相应的BNN吲哚产物8。

总结
Y. Kondo与M. Shigeno研究团队成功开发出一种选择偶氮苯作为起始原料,通过采用BBr3进行的邻位硼化以及采用Grignard试剂进行的二烷基化关环的两步反应过程,进而成功完成一系列BNN吲哚分子的构建。之后,作者通过对NICS值的计算表明,带有B-N-N键的五元杂环分子,具有中等程度的芳香性。同时,作者通过进一步的实验研究表明,硼化偶氮苯化合物的关环过程的反应机理主要涉及通过Grignard试剂对底物中偶氮基团的亲核加成与分子内关环,以及后续涉及B-Br键的亲核取代过程。
参考文献
[1] (a) P. G. Campbell, A. J. V. Marwitz, S. Liu, Angew. Chem. Int. Ed. 2012, 51, 6074. doi: 10.1002/anie.201200063.(b) C. R. McConnell, S. Liu, Chem. Soc. Rev. 2019, 48, 3436. doi: 10.1039/C9CS00218A.
(c) M. M. Morgan, W. E. Piers, Dalton Trans. 2016, 45, 5920. doi: 10.1039/C5DT03991F.
(d) Z. Liu, T. B. Marder, Angew. Chem. Int. Ed. 2008, 47, 242. doi: 10.1002/anie.200703535.
(e) Z. X. Giustra, S. Liu, J. Am. Chem. Soc. 2018, 140, 1184. doi: 10.1021/jacs.7b09446.
(f) G. E. Rudebusch, L. N. Zakharov, S. Liu, Angew. Chem. Int. Ed. 2013, 52,.316doi:10.1002/anie.201304443.
(g) S. Xu, L. N. Zakharov, S. Liu, J. Am. Chem. Soc. 2011, 133, 20152. doi: 10.1021/ja2097089.
(h) H. Braunschweig, A. Damme, J. O. C. Jimenez-Halla, B. Pfaffinger, K. Radacki, J. Wolf, Angew. Chem. Int. Ed. 2012, 51, 10034. doi: 10.1002/anie.201205795.
[2] (a) G. R. Humphrey, J. T. Kuethe, Chem. Rev. 2006, 106, 2875. doi: 10.1021/cr0505270.(b) M. Bandini, A. Eichholzer, Angew. Chem. Int. Ed. 2009, 48, 9608. doi: 10.1002/anie.200901843.
[3] (a) E. R. Abbey, S. Liu, Org. Biomol. Chem. 2013, 11, 2060. doi: 10.1039/C3OB27436E.(b) B. Su, R. Kinjo, Synthesis 2017, 49, 2985. doi: 10.1055/s-0036-1588832.
(c) L. Weber, Coord. Chem. Rev. 2008, 252, 1. doi: 10.1016/j.ccr.2007.02.014.
[4] (a) E. R. Abbey, L. N. Zakharov, S. Y. Liu, J. Am. Chem. Soc. 2010, 132,16340. doi: 10.1021/ja107312u.(b) E. R. Abbey, L. N. Zakharov, S. Liu, J. Am. Chem. Soc. 2011, 133, 11508. doi: 10.1021/ja205779b.
[5] C. Ma, J. Zhang, J. Li, C. Cui, Chem. Commun. 2015, 51, 5732. doi: 10.1039/C5CC00471C. [6] X. Wang, A. Narita, X. Feng, K. Müllen, J. Am. Chem. Soc. 2015, 137, 7668. doi: 10.1021/jacs.5b05056.. [7] (a) T. Hatakeyama, S. Hashimoto, S. Seki, M. S. Nakamura, J. Am. Chem. Soc. 2011, 133, 18614. doi: 10.1021/ja208950c.(b) L. Niu, H. Yang, R. Wang, H. Fu, Org. Lett. 2012, 14, 2618. doi: 10.1021/ol300950r.
[8] (a) N. Ishida, T. Moriya, T. Goya, M. Murakami, J. Org. Chem. 2010, 75, 8709. doi: 10.1021/jo101920p.(b) D. L. Crossley, I. A. Cade, E. R. Clark, A. Escande, M. J. Humphries, S. M. King, I. Vitorica-Yrezabal, M. J. Ingleson, M. L. Turner, Chem. Sci. 2015, 6, 5144. doi: 10.1039/C5SC01800E.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载












No comments yet.