
本文作者:杉杉
导读
近日,郑州大学杨贯羽和复旦大学黎占亭课题组合作共同在Green Chemistry上发表论文,使用天然可再生的低毒性没食子酸(gallic acid)作为有机催化剂,以水作为溶剂于室温下反应,即可实现2-氨基酚和2-羟基酚的氧化交叉缩合反应(aerobic oxidative cross-cyclocondensations),从而获得中等到较高收率的2-羟基-吩恶嗪(phenoxazin)-3-酮化合物。
Simple and Efficient Syntheses of 2-Hydroxy-3H-phenoxazin-3-ones by Aerobic Oxidative Cross-Cyclocondensation in Water
Wenhao Li,Wenxue Duan, Qingxuan Tang, Zhan-Ting Li* and Guanyu Yang*
Green Chem. ASAP DOI:10.1039/D0GC03861J
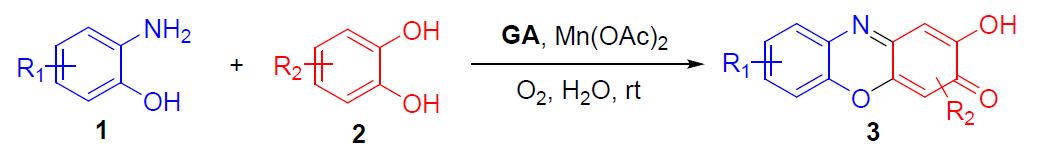
正文
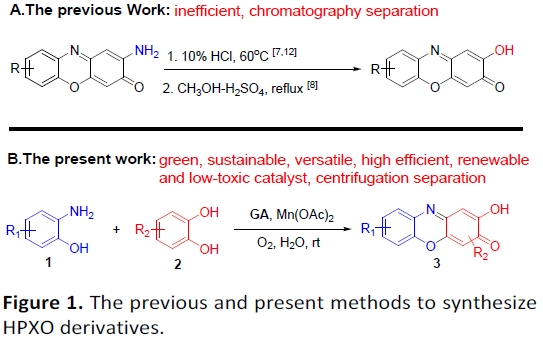
3H-吩恶嗪-3-酮(PXO)作为一种广泛的天然发色团,在古代被用作染料和色素,现代用于生物成像的探针。同时,PXO衍生物具有一定的生物活性,可用于治疗疟疾、癌症、炎症、细菌感染、神经退行性疾病等疾病。放线菌素D(Actinomycin D)是研究最广泛的PXO衍生物之一,具有2-氨基-PXO骨架,并具有抗生素和抗癌生物活性。眼黄素(Xanthommatin)是一种天然的2-氨基-PXO生物色素,在昆虫、蜘蛛和头足类动物中含量很高。前期的研究[1],主要集中在2-氨基-PXOs骨架的构建。Yoo和Rill等[2]发现以2-羟基-3H-苯恶嗪-3-酮(HPXO)为中心生色团结构对DNA的结合具有异常高的选择性。然而,关于HPXOs合成的报道很少[3]。如漆酶介导[4](laccasemediated)的3-氨基-2-羟基苯磺酸在甲醇/H2O中的均二聚反应,生成HPXO-1,6-二磺酸的收率很低,仅为4%。同时,在邻氨基苯酚与对苯醌的反应以及3-羟基邻氨基苯甲酸的氧化反应中也产生少量副产物HPXO[5-6]。此外,使用2-氨基-PXO进行羟基取代合成HPXO的常用方法需强酸性介质[2-3,7](Figure 1. A)。因此,仍需开发一种绿色环保且高效合成HPXO的方法。最近,本课题组开发了一种高效制备二硫醚的方法[8],该方法使用天然可再生,廉价且低毒的没食子酸(GA)作为有机催化剂,廉价的MnCO3作为助催化剂,氧气作为末端氧化剂,并在水中反应。在此,该课题组成功将此体系用于2-氨基酚(1)和2-羟基酚(2)的氧化交叉缩合反应,从而合成一系列HPXOs(3),这是以前没有报道的转化(Figure 1. B)。
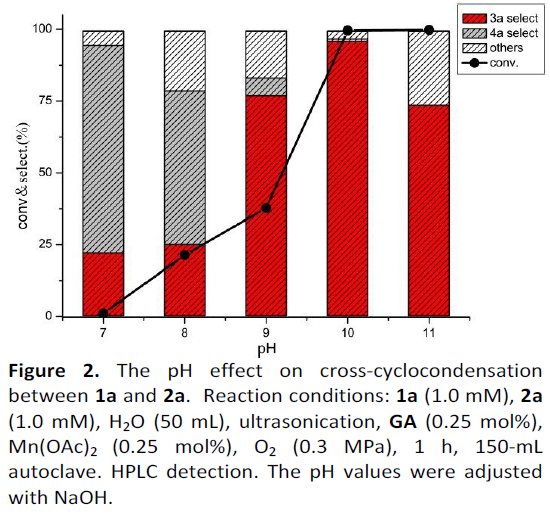
基于前期工作的总结[8],作者以2-氨基苯酚(1a)和2-羟基苯酚(2a)作为模型底物,以GA为催化剂,水溶性更高的Mn(OAc)2为助催化剂时,获得了48%的HPXO(3a),43%的2-氨基-PXO(4a)和9%其他产物,从而表明反应同时发生2a至3a的交叉环缩合和1a至4a的均二聚。随后,将反应物进行超声波处理,并用较低的反应物浓度筛选不同的pH值(Figure 2)。当pH从7.0变为11.0时,交叉环缩合产物的收率也有所变化。值得注意的是,在pH = 10.0时,可获得3a,HPLC收率为96.4%。但是,在pH = 11时,3a的收率下降,可能的原因是3a在强碱性条件下降解。
为了进一步了解反应的过程,作者进行了相关的对照实验(Scheme 1)。在pH为10时,2a的氧化对照实验中,约有一半的2a在20秒内转化为邻苯醌(Scheme 1, A)。在相同条件下,1a和2a的交叉缩合反应可得到高收率主产物3a和副产物4a。根据文献方法[9],将邻苯醌加入到2-氨基苯酚的水溶液中后,橙色3a立即沉淀出来,邻苯醌几乎消失了(Scheme 1, B)。此外,关于GA氧化的电子顺磁共振研究表明[10],GA形成其自由基随pH而变化,pH=13时自由基浓度比pH=9时高1000倍。因此,在较高pH下由GA形成的更多自由基会促进氧化,并且2a迅速转化为相应的邻苯并醌,这对随后1a中亲核性N原子的1,4-加成是有利(Scheme 1, C)。反应结果表明,交叉环缩合在0.5小时内完成并选择性地得到3a。值得注意的是,通过酸化、离心和重结晶的简单处理,克级获得98%收率的3a,进一步证明了反应的实用性。

在获得上述最佳反应条件后,作者开始对底物范围进行了扩展(Table 1)。反应结果表明,一系列含有给电子基团(如甲基和甲氧基)、吸电子基团(如氟、氯、溴、羧基和磺酸),均可在1h内完成反应,获得中等以上收率的HPXO产物3a–3z。同时,HPXO衍生物在药物和荧光探针中具有潜在的应用,水溶性基团的引入必不可少,通过此方案成功的引入一个或两个羧基或磺酸基的3s–3y,然而由于较高的水溶性,导致收率略低。
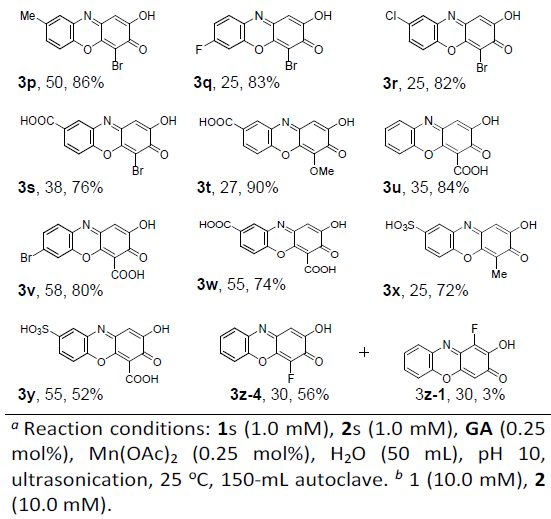
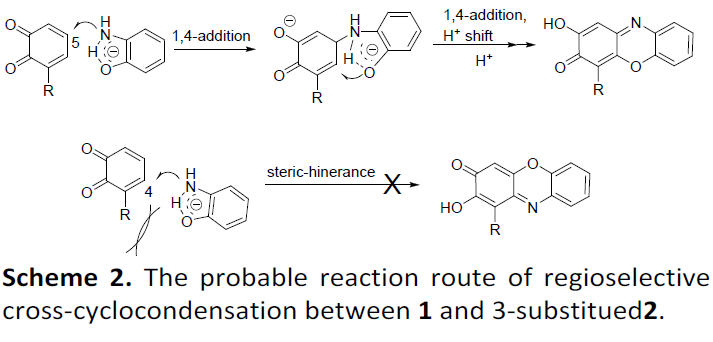
最后,作者提出了一种可能的反应机理(Scheme 2)。首先,在碱性条件下,2-氨基苯酚经去质子化形成阴离子,在N和O原子之间具有分子间氢键。其次,中间体邻苯并醌具有两个迈克尔加成受体,可经连续两次迈克尔加成(即邻苯甲醌的5C位点上的分子间氮杂-迈克尔加成,4C位点的分子内氧杂-迈克尔加成),从而形成所需的目标产物。值得注意的是,当使用3-取代的2-羟基苯酚时,在5C位点的氮杂-迈克尔加成生成4-取代羟基吩恶嗪酮产物,而在4C位点则生成1-取代的产物。然而,由于取代基在2的3C位上的空间位阻可完全抑制在4C位上的氮杂-迈克尔加成,因此交叉环缩合具有优异的区域选择性(3d–3y)。相反,较小的F取代不能抑制在4C位置发生氮杂-迈克尔加成反应,因此3-F-2-羟基苯酚与1a的反应生成3z-4及其位置异构体3z-1的混合产物。此外, 1a与2,3-二羟基苯甲酸或其酯的反应不能提供所需的HPXO,而是4a作为主要产物,这可能是因为更大的羧基空间位阻可防止交叉环缩合。然而,通过2-氨基苯酚和3-氰基-2-羟基苯酚之间的反应可以获得3u–3w和3y,其中氰基被完全氧化水解为羧基。
总结
郑州大学杨贯羽和复旦大学黎占亭课题组合作报道了一种新颖、高效且通用的合成2-羟基-吩恶嗪-3-酮化合物方法,即以天然可再生的低毒性没食子酸(GA)作为有机催化剂,Mn(OAc)2为助催化剂,氧气为末端氧化剂,水为溶剂。同时,该反应具有高收率、温和的反应条件、操作简单等优点。

















No comments yet.