
本文作者:杉杉
导读
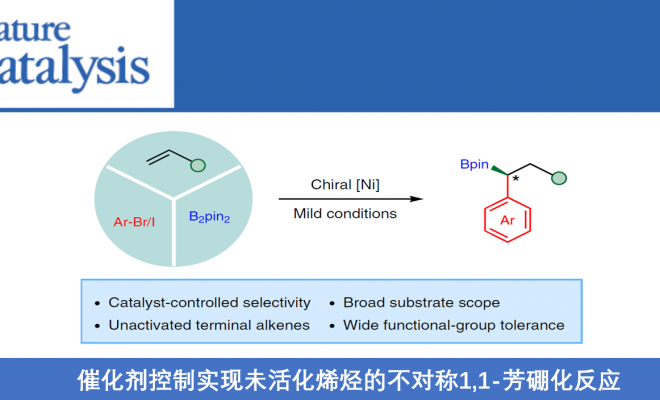
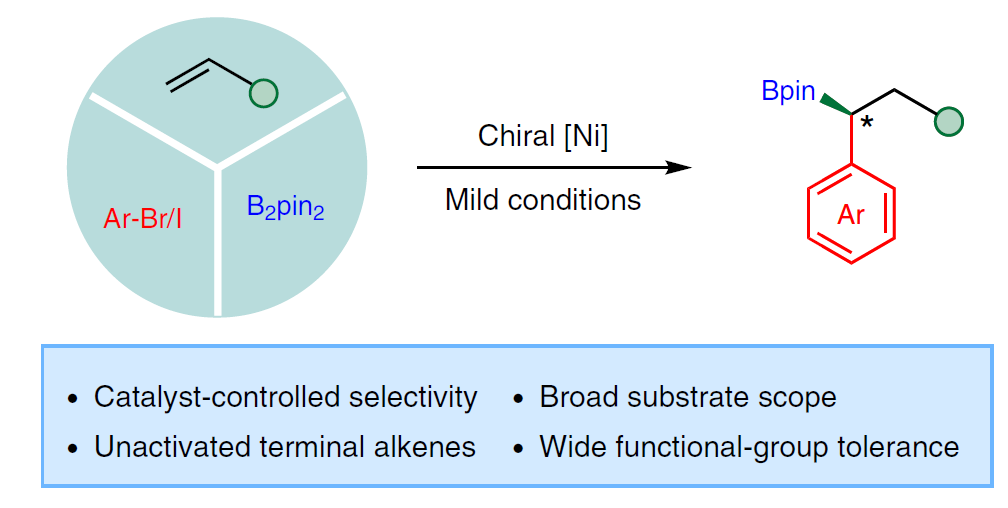
烯烃的对映选择性双官能化,是从简单原料合成复杂手性分子的有效策略。近日,武汉大学阴国印课题组在Nat. Catal.上发表论文,报道了镍催化实现末端烯烃(未活化)分子间的对映选择性1,1-芳硼化反应,具有高区域选择性和对映选择性(镍催化剂决定)。同时,烯丙基苯底物也同样取得良好的结果。此外,通过对硼酸酯产物的衍生化,进一步证明了反应的实用性。
Catalyst-controlled enantioselective 1,1-arylboration of unactivated olefins
Wang Wang, Chao Ding and Guoyin Yin*
Nat. Catal. 2020, 3, 951-958. DOI:10.1038/s41929-020-00523-8 https://doi.org/10.1038/s41929-020-00523-8
正文
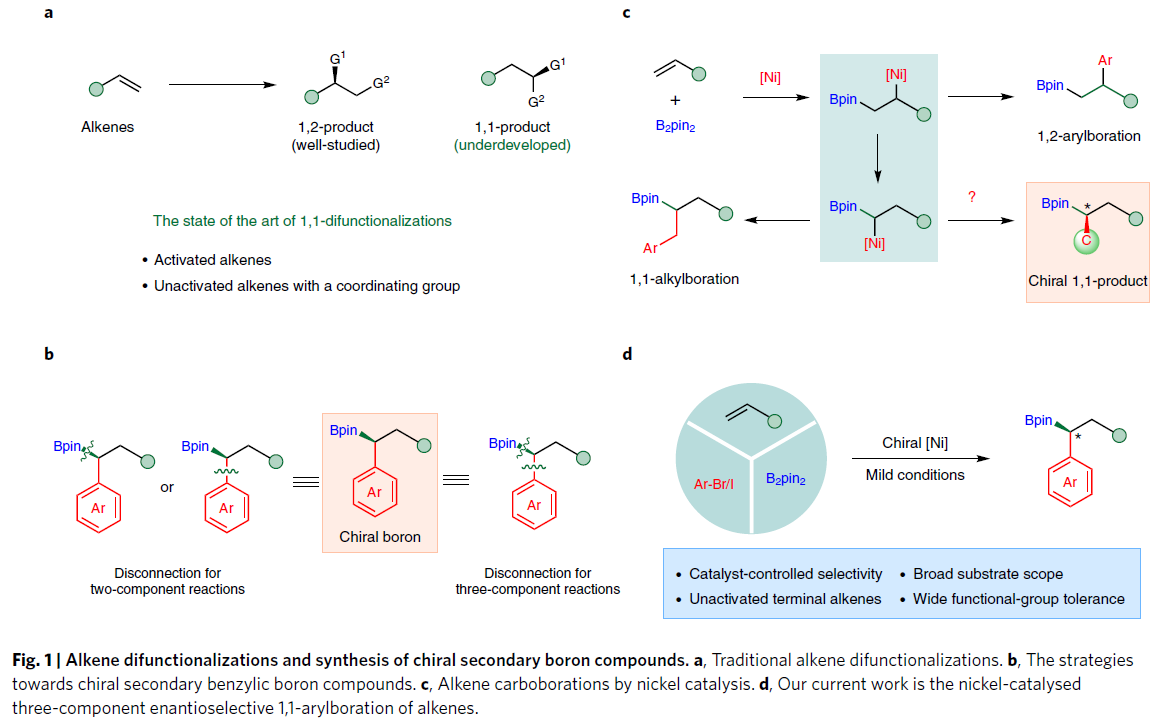
烯烃作为常见的工业原料,具有易得且成本低的优点,其选择性转化也一直是有机合成界的研究课题。烯烃的对映选择性双官能化,是快速构建复杂手性分子的有效策略。而过渡金属催化策略,不仅实现1,2-区域选择性,同时也为1,1-区域选择性提供了机会。然而,对于不对称1,1-双官能化反应的研究仍然不完善,同时,烯烃的范围限于活化的烯烃和带有配位基团的烯烃,而实现没有导向基团的未活化烯烃的1,1-双官能化反应仍具有巨大的挑战。
手性仲苄基硼酸酯作为通用的合成子,广泛应用于现代有机合成中。不对称的两组分交叉偶联反应,作为一种高效的方法,可快速合成此类化合物。如Morken课题组[1]报道了偕-二硼试剂与芳基底物的交叉偶联,Fu[2]和Watson[3-4]课题组报道了苄基亲电试剂与硼试剂的交叉偶联。此外,Yun[5]和Lu[6]课题组报道了烯基芳烃的不对称硼氢化反应,Zhou[7]课题组报道了不对称苄基卡宾插入BH3中的反应。相反,通过廉价且易得的起始原料,实现三组分的1,1-双官能化策略,可快速合成手性硼化合物。Toste[8]和Brown[9]课题组利用钯或钯/铜催化实现此类反应,但烯烃的范围有限。
烯烃的碳硼化(carboboration)反应,是一种快速合成烷基硼酸酯的有效方法。最近,Brown[10-13]和Yin[14-15]课题组报道了镍催化碳硼化反应,涉及由烯烃迁移插入Ni-B键中,从而形成烷基镍中间体。如果该中间体直接与芳基卤化物反应,则获得1,2-芳基硼酸酯产物。而使用苄基卤化物时,则获得1,1-烷基硼化产物,从而为合成手性硼化合物提供新的可能(Fig. 1)。在此,武汉大学阴国印课题组报道了通过镍催化,实现未活化烯烃的不对称1,1-芳硼化反应,从而获得1,1-双官能化产物,具有良好的收率和对映选择性。此外,烯丙基苯衍生物也在相同条件下,以优异的区域选择性获得仲烷基硼化合物。
首先,作者以末端烯烃(1),4-溴苯甲酸甲酯(2)和B2pin2(3)作为模型底物,进行了相关不对称1,1-双官能化反应的条件筛选(Table 1)。反应结果表明,当使用由氯化镍和L7制备的镍催化剂rac-[Ni]-1,可获得87%收率的目标产物4。

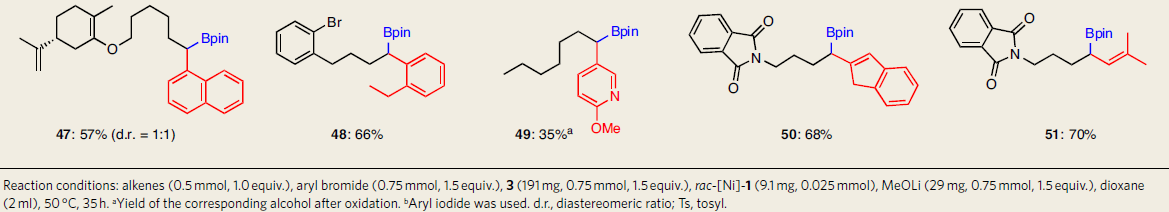
在获得上述最佳反应条件后,作者开始对未活化的单取代烯烃和芳基卤化物的底物范围进行了扩展(Table 2)。1-庚烯可与几种芳基溴底物反应,获得中等收率的产物6-7、32和40,表明区域选择性是由催化剂决定的。而在与6-溴-1-己烯的反应中未检测到环化产物(11)。伯烷基溴化物(11和39)、烷基(12)和芳基(17)甲苯磺酸盐、芳基氯化物(13、26、27和43)和二取代的烯烃(47),均与体系兼容,表明该反应具有良好的化学选择性。除芳基亲电试剂外,烯基亲电试剂也可顺利地进行芳硼化反应,得到相应的产物(50和51)。此外,含有酯、酮、胺、酰亚胺、碳酰胺、环氧基和硅烷基团以及杂环(如吡咯、苯并噻吩、噻吩、官能化的吲哚和吡啶)时,均与体系相容,从而说明反应具有良好的官能团耐受性。值得注意的是,在所有情况下均未检测到相关的1,2-和2,1-异构体。
紧接着,作者以烯丙基苯及其衍生物作为末端烯烃底物,进行了相关的扩展(Table 3)。值得注意的是,若反应是通过碳-Heck型反应引发,则会得到两种异构体的混合物(53和54)。因此,镍催化的芳硼化反应是通过硼基-Heck型反应,而不是碳-Heck型反应引发的。
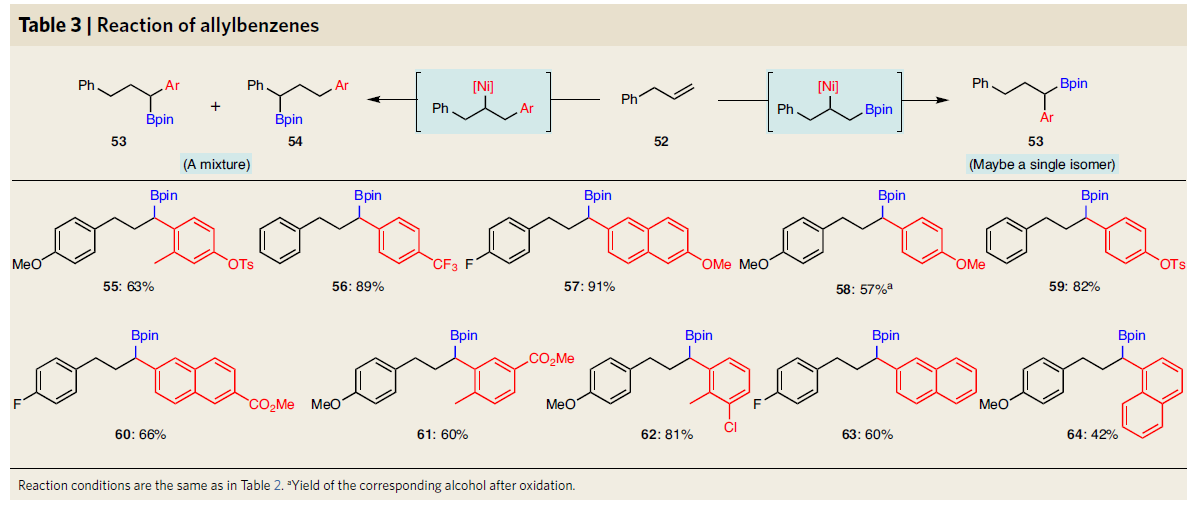
随后,作者使用手性镍催化剂([Ni]-1*)对反应进行了不对称合成(Table 4)。未活化的α-烯烃和芳基卤化物,均可获得相应的手性烷基硼化合物。同时,反应具有良好的官能团耐受性。此外,在空间上受阻的芳基卤化物以及杂环芳基溴化物,均与体系兼容。值得注意的是,克级反应(70和87)获得相同水平的对映选择性,从而证明了该反应的可扩展性。

此外,气态烯烃(乙烯和丙烯)也同样可以获得相应的1,1-芳基硼酸酯化产物95-97,但与乙烯产物的e.e偏低(Fig. 2a)。而具有较长碳链的烯烃具有更好的对映选择性(72),表明适当增加烯烃的位阻可提高对映选择性。

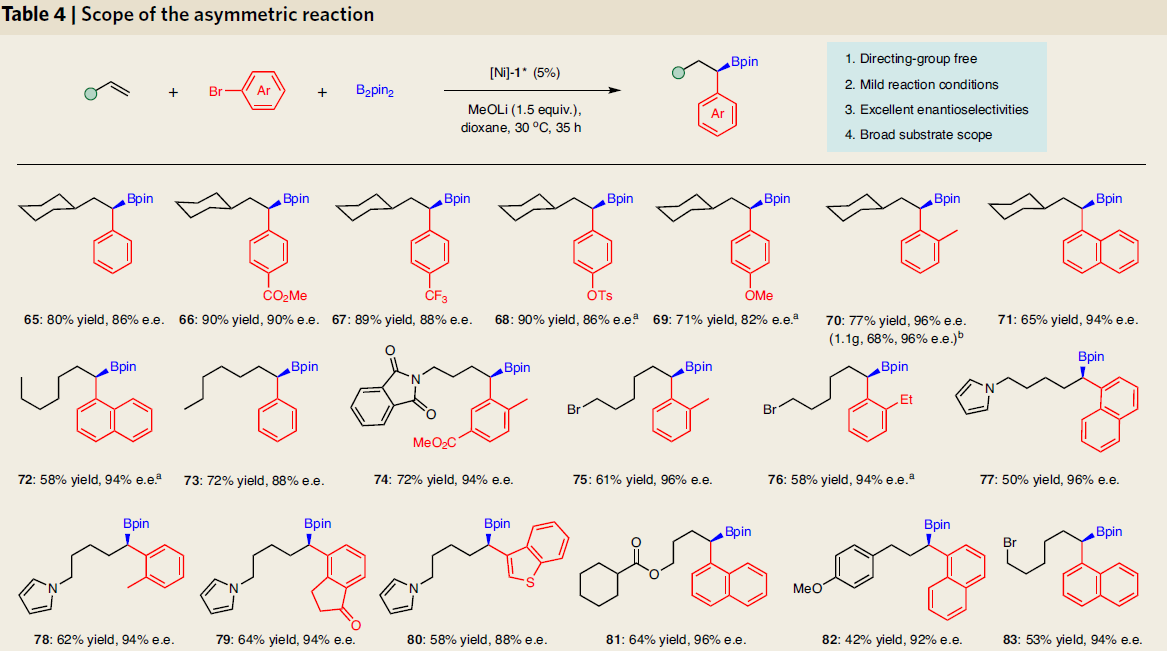
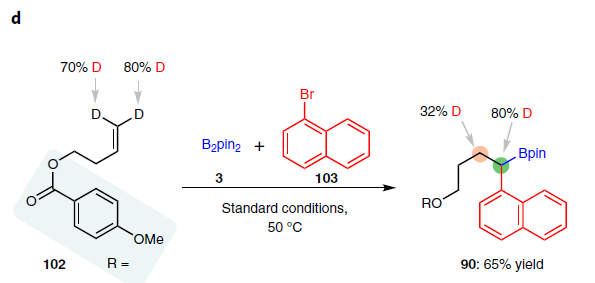
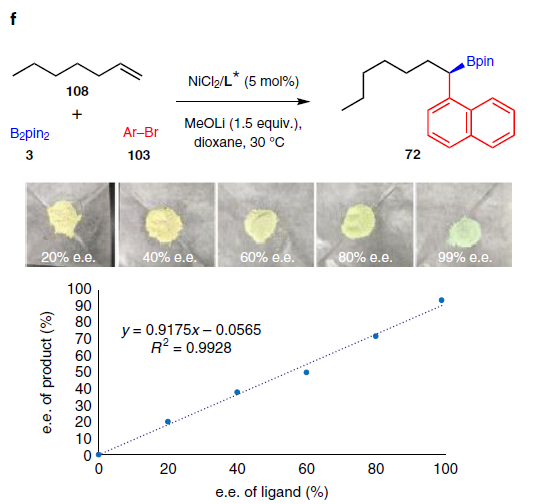
为了进一步了解反应的机理,作者进行了相关的对照实验。首先,在标准条件下,制备了两种可能的中间体(98和99),并与芳基溴100进行反应,然而,均未发现Suzuki-Miyaura产物(56),该结果表明1,1-和1,2-二硼基化合物都不是反应的中间体(Fig. 2b)。其次,在标准条件下加入乙烯基硼化合物101时,未检测到产物56,这表明在迁移过程中烯烃中间体不太可能从镍配合物中解离(Fig. 2c)。而末端氘标记的烯烃(102)迁移到相邻碳原子中的产物,这表明该反应涉及1,2-镍迁移(Fig. 2d)。当使用带有芳基溴化物的烯烃(104)时,在反应中未检测到分子内环化产物。若加入邻位取代的芳基溴化物105时,仅分离出分子间三组分产物48(Fig. 2e)。值得注意的是,这些结果与Brown’s无配体体系不一致[16]。最后,配体的e.e.与产物的e.e具有线性关系,显示了对映选择性测定步骤可能涉及单个二胺配体和一个镍物种(Fig. 2f)。
基于相关文献[17-18]以及上述的结果,作者提出了可能的反应机理(Fig. 2g)。首先,[Ni]II催化剂通过归中反应产生[Ni]I配合物(I),再与B2pin2经转金属化,从而生成[Ni]I-Bpin配合物(II)。紧接着,烯烃迁移插入形成烷基-[Ni]I中间体(III),经再1,2-金属迁移形成稳定的中间体IV。随后,中间体IV与芳基溴发生氧化加成生成[Ni]III中间体V。最后,V经还原消除生成手性硼产物,并再生[Ni]I催化剂。

为了进一步证明反应的实用性,作者进行了相关的衍生化实验(Fig. 3)。手性硼化合物,若经氧化则形成相应的手性苄醇109,若经Matteson反应则形成110-112,若经一系列的氧化和环化反应则形成手性七元环醚113。
总结
武汉大学阴国印课题组报道了镍催化未活化烯烃的不对称1,1-芳硼化反应。同时,可从易得的末端烯烃,市售的芳基卤化物和二硼化试剂,直接合成具有价值的对映体富集的仲苄基硼酸酯化合物。此外,通过相关后期的衍生化,进一步证明了反应的实用性。
参考文献
1. Sun, C., Potter, B. &Morken, J. P. A catalytic enantiotopic-group-selective Suzuki reaction for the construction of chiral organoboronates. J. Am. Chem. Soc. 136, 6534–6537 (2014).
2. Wang, Z., Bachman, S., Dudnik, A. S. & Fu, G. C. Nickel-catalyzed enantioconvergentborylation of racemic secondary benzylic electrophiles. Angew.Chem. Int. Ed. 57, 14529–14532 (2018).
3. Basch, C. H., Cobb, K. M. & Watson, M. P. Nickel-catalyzed borylation of benzylic ammonium salts: stereospecific synthesis of enantioenriched benzylic boronates. Org. Lett. 18, 136–139 (2016).
4. Pound, S. M. & Watson, M. P. Asymmetric synthesis via stereospecific C–N and C–O bond activation of alkyl amine and alcohol derivatives. Chem. Commun. 54, 12286–12301 (2018).
5. Noh, D., Chea, H., Ju, J. & Yun, J. Highly regio- and enantioselective copper-catalyzed hydroboration of styrenes. Angew. Chem. Int. Ed. 48, 6062–6064 (2009).
6. Chen, X., Cheng, Z., Guo, J. & Lu, Z. Asymmetric remote C–H borylation of internal alkenes via alkene isomerization. Nat. Commun. 9, 3939 (2018).
7. Pang, Y. et al. Rhodium-catalyzed B−H bond insertion reactions of unstabilized diazo compounds generated in situ from tosylhydrazones. J. Am. Chem. Soc. 140, 10663–10668 (2018).
8. Nelson, H. M., Williams, B. D., Miro, J. &Toste, F. D. Enantioselective 1,1-arylborylation of alkenes: merging chiral anion phase transfer with Pd catalysis. J. Am. Chem. Soc. 137, 3213–3216 (2015).
9. Bergmann, A. M., Dorn, S. K., Smith, K. B., Logan, K. M. & Brown, M. K. Catalyst-controlled 1,2-and 1,1-arylboration of α-alkyl alkenyl arenes. Angew. Chem. Int. Ed. 58, 1719–1723 (2019).
10. Logan, K. M., Sardini, S. R., White, S. D. & Brown, M. K. Nickel-catalyzed stereoselectivearylboration of unactivated alkenes. J. Am. Chem. Soc. 140, 159–162 (2018).
11. Logan, K. M., Smith, K. B. & Brown, M. K. Copper/palladium synergistic catalysis for the syn- and anti-selective carboboration of alkenes. Angew. Chem. Int. Ed. 54, 5228–5231 (2015).
12. Chen, L.-A., Lear, A. R., Gao, P. & Brown, M. K. Nickel-catalyzed arylboration of alkenylarenes: synthesis of boron substituted quaternary carbons and regiodivergent reactions. Angew. Chem. Int. Ed. 58, 10956–10960 (2019).
13. Joung, S., Bergmann, A. M. & Brown, M. K. Ni-catalyzed 1,2-benzylboration of 1,2-disubstituted unactivated alkenes. Chem. Sci. 10, 10944–10947 (2019).
14. Wang, W. et al. Migratory arylboration of unactivated alkenes enabled by nickel catalysis. Angew.Chem. Int. Ed. 58, 4612–4616 (2019).
15. Wang, W., Ding, C., Pang, H. & Yin, G. Nickel-catalyzed 1,2-arylboration of vinylarenes. Org. Lett. 21, 3968–3971 (2019).
16. Sardini, S. R. et al. Ni-catalyzed arylboration of unactivated alkenes: scope and mechanistic studies. J. Am. Chem. Soc. 141, 9391-9400 (2019).
17. Chen, L.-A., Lear, A. R., Gao, P. & Brown, M. K. Nickel-catalyzed arylboration of alkenylarenes: synthesis of boron substituted quaternary carbons and regiodivergent reactions. Angew. Chem. Int. Ed. 58, 10956–10960 (2019).
18. Li, Y. et al. Nickel-catalyzed 1,1-alkylboration of electronically unbiased terminal alkenes. Angew.Chem. Int. Ed. 58, 8872-8876 (2019).











No comments yet.