
译自Chem-Station网站日本版 原文链接:計算化学:DFTって何? PartIII
翻译:炸鸡
在上一回《计算化学的一点简单科普(3):从被嫌弃到被喜欢的DFT》中,笔者简要介绍了1930年-1980年之间量子化学的发展史。
这一期笔者将介绍DFT计算中使用的主要泛函。
读完前一期和这一期的内容,相信读者们都会对DFT计算的历史背景,理论,不足点,泛函的种类和Gaussian软件的计算流程有一个更加深刻细致的理解。当别人问你什么是DFT计算时,你也能自信地回答了。
上回没有言及的量子化学在1930年-1980年之间的进展还有基函数的开发(1950年Boys),罗特汉方程的提出(1950年Roothaan, Hall)。还有对现代计算化学极其重要的Gaussian软件的开发(1970年、Pople),Gamess软件的开发(1982年)。
各种各样的泛函
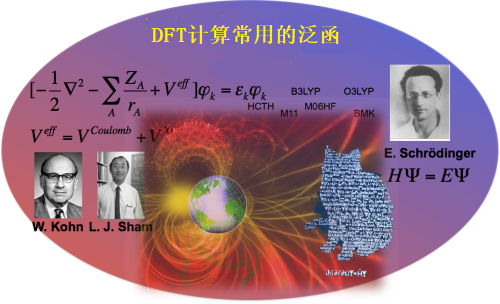
在《计算化学的一点简单科普(1):什么是泛函?》中曾提到过,交换关联泛函是Kohn-Sham方程中唯一取近似的部分。所以DFT计算结果的准确性取决于它选择的泛函。虽然迄今为止有很多泛函被开发出来,这些泛函可以分为以下几类:
局部密度近似(LDA)泛函:Local Density Approximation (LDA) Functional
广义梯度近似(GGA)泛函:Generalized Gradient Approximation (GGA) Functional
Meta-GGA泛函:Meta-Generalized Gradient Approximation (Meta-GGA) Functional
混合泛函(杂化泛函):Hybrid Functional
半经验泛函:Semi-Empirical Functional
渐进泛函:Progressive Functional
和有固定方程的薛定谔方程不同,交换关联泛函没有一个最优的形式,每位计算化学家都依照自己的研究需要,选择自己的参数制作属于自己的泛函。
可以根据交换关联泛函使用的变量来分类,比如有和电子密度成比例的交换相关泛函,还有和电子密度的微分成比例的交换相关泛函。更复杂的是,还有人用几个泛函组合起来拼接成杂化泛函。
今天将为读者介绍有机合成化学家经常使用的两个具有代表性的泛函。
B3LYP
B3LYP是最早的杂化泛函,是有机化学学界最常使用的泛函。于1993年由Becke提出。
这个泛函不仅包含Hartree-Fock交换积分的混合比例,还有B88交换泛函、LYP相关泛函以及LDA交换相关泛函之间的差按三个参数比例的加和。
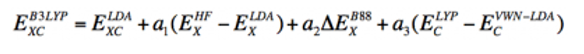
半经验的三个参数分别为a1 = 0.2、a2 = 0.72、a3 = 0.81。这个三个参数是不能随意更改的基准,经过无数的实验证实这三个值能让计算值接近实验值,并不是经过数学公式推导出来的。但请不要认为建立在这个“经验“参数上的DFT计算结果就是不正确的,不如这样想——比起如何更准确求得薛定谔方程的近似解,用DFT算出的结果比MP2要更接近于实验结果。
“人们为什么要用计算化学?”
如果计算结果和实验结果一致,实验结果的正确性就得到了佐证,但反过来说无论计算结果从数学上推导是多么地正确,一旦与实验结果存在较大差异,那么就没有人会使用。类似的情况也适用于酶、基因序列等相似性搜索。
在检索酵素基因序列时,我们大脑里都会有这样一个假设
“功能类似的蛋白质,序列也一定类似”
这个假设凭直觉来想没有什么问题,但实质上没有经过严密的数学证明。但为什么很多科学家都遵循这个假设,用相似性检索呢?因为实验结果证实这个假设具有合理性!!同样,DFT计算结果也是经过实验验证的。
M06系泛函

M06泛函是我最近经常用的,这个泛函由美国量子化学家Donald Gene Truhlar开发。M06属于半经验泛函,有高达38个半经验参数。但我相信M06不会一直是学术界主要用的泛函,M06这样繁复的泛函只是真正精炼的泛函被开发出来之前的权宜之计。
我脑海里一直有个原则,即在含有过渡金属的体系中通常使用M06,而在没有过渡金属的体系中则常用M06-2x。此外,由于它是一种可以估算弱相互作用的通用泛函,因此在像周环反应等情境下,经常看到先使用B3LYP进行结构优化,然后再用M062X进行单点计算的例子。
实际计算流程
计算机软件变得越来越便于用户使用了,即使是零基础小白也能算出化合物的能量,优化化合物的结构,模拟谱图。计算机软件的新手友好程度堪比游戏了。
制作输入文件并进行计算,得到结果。然而,要判断这些结果是否正确,具有何种意义,就需要有机化学知识。此外,为了在计算报错时做出正确处理,还需要深入了解背后的理论和计算程序。
计算化学虽然被称为计算,但能够理解计算机实际进行了哪些计算工作的人并不多。例如,分子结构最优化的计算过程大致如下:
1.读取坐标信息
2.能量计算(Roothaan方程和SCF计算)
3.力计算
4.结构最优化
查看计算结束后的后缀名为log.的文件,您可以了解哪个部分是限速的,以及更改哪些参数可以提高计算速度等。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.

















No comments yet.